CDRH COVID-19 Serology/Antibody Templates - Serology Template
0910-0595_CDRH_serology-template.docx
Authorization of Medical Products for Use Emergencies
CDRH COVID-19 Serology/Antibody Templates - Serology Template
OMB: 0910-0595
OMB: 0910-0595
Exp. date 9/30/2025
Contains Nonbinding Recommendations
Serology Template for Test Developers1
This template provides the Food and Drug Administration’s (FDA) current recommendations concerning what data and information should be submitted to FDA in support of a pre-Emergency Use Authorization (EUA) submission/EUA request for a SARS-CoV-2 serology (antibody) test. FDA generally recommends that the following validation studies be conducted for SARS-CoV-2 serology tests: cross-reactivity/analytical specificity, class specificity, and clinical agreement. Additionally, in Section J.4 below, FDA also recommends a matrix equivalency study (as applicable) be conducted for SARS-CoV-2 serology tests, and, for quantitative tests, in Section J.6, FDA recommends the following additional validation studies be conducted: limit of blank (LoB), limit of detection (LoD), limit of quantitation (LoQ), linearity, precision, interference, and traceability, including certified reference material (CRM) accuracy and cutoff studies in the CRM units.
As described in the FDA guidance document: Policy for Coronavirus Disease-2019 Tests During the Public Health Emergency (Revised),2 FDA is providing recommendations in this and other EUA templates regarding testing that should be performed to ensure appropriate analytical and clinical validity, including descriptions of appropriate comparators, for different types of tests.
The EUA templates3 are intended to help test developers provide recommended validation data and other information to FDA, but alternative approaches can be used. This template reflects FDA’s current thinking on the topic, and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word should, means that something is suggested or recommended, but not required. For more information about EUAs in general, please see the FDA guidance document: Emergency Use Authorization of Medical Products and Related Authorities.4
Test developers interested in pursuing an EUA may submit a pre-EUA to begin discussions with the FDA or may submit an EUA request to covid19dx@fda.hhs.gov.
GENERAL INFORMATION ABOUT THIS TEMPLATE
Text highlighted in yellow [Text] should be completed by test developers as applicable to their specific test. Text in bold outlines the FDA’s additional recommendations for the developers’ consideration when completing the suggested information in each section.
Not all portions of this template may be relevant for all developers/tests. FDA recommends developers complete all portions that are relevant to facilitate a streamlined review.
This template is intended for tests indicated for the qualitative or quantitative detection of antibodies to SARS-CoV-2. If you are developing a test indicated for the detection of or correlation to neutralizing antibodies, please see the Template for Test Developers of Serology Tests that Detect or Correlate to Neutralizing Antibodies.5
Please be reminded that tests for the detection of antibodies against SARS-CoV-2 should not be distributed and/or used to diagnose current infection.
A test authorized under an EUA is only authorized for emergency use while the EUA is in effect.
We plan to update the template as appropriate as we learn more about COVID-19 and gain experience with the EUA process for these kinds of tests.
A developer that has provided data to the FDA may grant a right of reference to other developers, either broadly or to individual developers, to leverage that data. A right of reference provides a developer the ability to rely upon, and otherwise use, existing information in one regulatory submission for the purpose of supporting a different regulatory submission. In these cases, if the data is applicable to the new developer's test, the new developer may not have to repeat that validation for its submission to the FDA or FDA may recommend only a bridging study. Any developer seeking to leverage data regarding another developer’s EUA-authorized assay must obtain a right of reference from that developer.
EXAMPLE TEMPLATE:
PURPOSE FOR SUBMISSION
Emergency Use Authorization (EUA) request for distribution and/or use of the [test name] in [indicate where the test would be performed - e.g., laboratories certified under the Clinical Laboratory Improvement Amendments of 1988 (CLIA), 42 U.S.C. §263a, to perform high/high or moderate/high, moderate or waived complexity (for point of care use), etc.], for the [qualitative, or quantitative] detection of [specify types of antibodies e.g., IgM, IgG, total] antibodies to SARS-CoV-2 in [specify matrices.]
MEASURAND
[Specify if the test is a qualitative or a quantitative test, what antibodies the test detects and whether it can differentiate between IgM and IgG or if it detects total antibody without differentiation.]
APPLICANT
[Official name, address, and contact information (including phone number and email address) of applicant and primary correspondent.]
PROPRIETARY AND ESTABLISHED NAMES
Proprietary Name: [test name]
Established Name: [test name]
REGULATORY INFORMATION
Approval/Clearance Status:
The [test name] is not approved, cleared or subject to an approved investigational device exemption.
[If the test has been previously reviewed in an EUA request or pre-EUA submission, please provide the submission number.]
Panel Code: MI for Microbiology tests
Review Group: Division of Microbiology Devices/VIR2
Product Code:
QKO - reagent, coronavirus serological
PROPOSED INTENDED USE
Intended Use (IU):
The proposed IU will be finalized based on, among other things, the data provided and recommendations from public health authorities at the time of authorization. Example text is provided below but may be adapted according to the specific emergency situation addressed by the device, proposed intended use population, testing sites, or performance characteristics.
The [test name] is a [specify test technology e.g., Enzyme-Linked Immunosorbent Assay (ELISA)] intended for [qualitative or quantitative] detection of [specify the antibody class or classes that are being detected or indicate whether the test only detects total antibodies] antibodies to SARS-CoV-2 in human [specify matrices including anticoagulants]. The [test name] is intended for use as an aid in identifying individuals with an adaptive immune response to SARS-CoV-2, indicating recent or prior infection. The [test name] should not be used to diagnose or exclude acute SARS-CoV-2 infection. At this time, it is unknown for how long antibodies persist following infection and if the presence of antibodies confers protective immunity. Testing is limited to laboratories certified under the Clinical Laboratory Improvement Amendments of 1988 (CLIA), 42 U.S.C. § 263a, that meet the requirements to perform [insert testing complexity, e.g., moderate complexity, high complexity, or waived tests. This test is authorized for use at the Point of Care (POC), i.e., in patient care settings operating under a CLIA Certificate of Waiver, Certificate of Compliance, or Certificate of Accreditation.].
Results are for the detection of [specify antibodies detected] SARS CoV-2 antibodies. [Specify antibodies detected] antibodies to SARS-CoV-2 are generally detectable in blood several days after initial infection, although the duration of time antibodies are present post-infection is not well characterized. Individuals may have detectable virus present for several weeks following seroconversion.
Laboratories within the United States and its territories are required to report all results to the appropriate public health authorities.
As applicable: The sensitivity of [test name] early after infection is unknown.
Negative results do not preclude acute SARS-CoV-2 infection. If acute infection is suspected, direct testing for SARS-CoV-2 is necessary.
False positive results for [test name] may occur due to cross-reactivity from pre-existing antibodies or other possible causes. [For lateral flow devices: Due to the risk of false positive results, confirmation of positive results should be considered using a second, different [as appropriate, IgG or IgM] assay].
The [test name] is only for use under the Food and Drug Administration’s Emergency Use Authorization.
Special Conditions for Use Statements:
For prescription use only
For in vitro diagnostic use
For Emergency Use Authorization only
Special Instrument Requirements:
The [test name] test is to be used with the [list all instruments, software, other applicable instrumentation, etc.].
If your test system includes an instrument, the instrumentation manual should be submitted as part of the EUA request. If your test system includes an instrument that was not previously cleared, approved, or authorized by FDA, please see additional discussion in the Product Manufacturing section and note that additional labeling information may be discussed during the EUA review.
DEVICE DESCRIPTION AND TEST PRINCIPLE
Please provide a device description. Example text has been added under each of the sub-headings. Please modify the example text as appropriate for tests that use a different test principle. For new technologies, FDA may request additional detailed information so we can adequately assess the known and potential risks and benefits associated with the device.
Product Overview/Test Principle:
[Describe the technology of the test and how this technology works to identify measurand (i.e., the test principle), the instruments/reader employed/required to perform the test from sample collection to result, and the sample types for which you claim to have specific performance characteristics, as described below. Please indicate if the test uses biotin-Streptavidin/avidin chemistry in any of the steps for coupling reagents.]
The [test name] uses the following: [List the antigen(s) and antibodies used in the assay to detect the antibodies in human samples.]
Description of Test Steps:
[List and describe in detail all of the steps of the test sequentially, from sample collection to assay report.]
[Step one]
[Step two]
[Etc.]
Control Material(s):
[List all control materials (provided with the test kit and/or required but not provided with the test kit, e.g., sold as a separate kit) and describe what they are, how they are expected to work, where in the testing process they are used, and the frequency of use. These controls should also be validated within your analytical and clinical studies described below in Section J.]
Controls that will be provided with the test kit include:
An external positive control for each antibody class detected (e.g., IgG, IgM) is needed to [describe need] and is used [describe use – please specify the composition and analyte level (with units, as applicable) of the positive control relative to the cut-off of your test (note that ideally the positive control concentration should be such that it is close to the cut-off of your test) and specify frequency of use.] The positive control should be consistent with the use of your device. For example, if you have a test that detects IgG antibodies, the positive control should contain IgG antibodies.
An external negative control is needed to [describe need] and is used [describe use – please specify the composition of the negative control and specify frequency of use].
A [other (e.g., sample adequacy, internal, etc.)] control is needed to [describe need] and is used [describe use – please specify the composition of the control and specify frequency of use].
Controls that are required but not provided with the test kit include [describe controls for purchase that you, the applicant, make available]. This/these control(s) is/are needed to [describe need] and is/are used [describe use – please also specify frequency of use].
Please note that any control used with your device (provided with the kit or not) should be validated in the context of your analytical and clinical studies (i.e., your studies should include use of these controls). External control materials are considered particularly important when good manufacturing practice (GMP) requirements are waived and reagent stability studies are limited.
Calibrator Material (as applicable):
[List all calibrator materials (provided with the test kit) and specify the composition and analyte level, with units, of each calibrator relative to the cut-off of the test and to the lower and upper limit of a measuring interval (for quantitative tests), how they are expected to work, where in the testing process they are used, and the frequency of use.]
Please note that external control materials should not be used as calibrators because calibrators are used to establish the candidate test result and the external control materials should be independent.
[For quantitative tests, describe the calibration curve, including: the number of calibrators, the mathematical model used to generate the calibration curve including the number of parameters; for each positive calibrator, identify the analyte, including the origin of the material (e.g., recombinant antibody, native samples, etc.); and for each calibrator, including zero calibrator, identify the matrix (e.g., serum or plasma from patient, buffer).]
[For quantitative tests, clearly describe how values were assigned based on an international/national standard, or CRM, including: information about the CRM, including issuing authority (e.g., World Health Organization (WHO), National Institute for Standards and Technology (NIST)), and units (e.g., binding antibody units (BAU)/mL for the WHO CRM), traceability of the test results to a CRM (the complete calibration hierarchy should be provided).] Please refer to International Organization for Standardization (ISO) 17511 “In vitro diagnostic medical devices- Requirements for establishing metrological traceability of values assigned to calibrators, trueness control materials and human samples” for recommendations on appropriate traceability establishment and documentation. [For each positive calibrator, you should provide assigned values (e.g., in BAU/mL) based on the calibration hierarchy to the CRM. You should describe how the highest calibrator value is related to the upper limit of the analytical measuring interval and how the lowest positive calibrator value is related to the lower limit of the analytical measuring interval.]
Test Result Reporting:
All test results are to be reported to healthcare providers and relevant public health authorities in accordance with local, state, and federal requirements, using appropriate LOINC and SNOMED codes, as defined by the Laboratory In Vitro Diagnostics (LIVD) Test Code Mapping for SARS-CoV-2 Tests6 provided by CDC. Core diagnostic data elements7 are to be collected for all tests, which have been defined by the Department of Health and Human Services (HHS), along with technical specifications for implementation for lab-based8 and non-lab-based9 tests.
INTERPRETATION OF RESULTS
All test controls should be examined prior to interpretation of patient results. If the controls are not valid, the patient results cannot be interpreted. [Appropriate control interpretation criteria should appear in your product labeling. If the test result involves the use of an algorithm/calculation when determining the final patient test result, please include a detailed description and any additional calibration materials that may be required.]
For assays with qualitative results: [Clearly describe how results are to be interpreted. If applicable, clearly indicate how to interpret numeric test values (e.g., Index Values) as positive or negative for the presence of antibodies against SARS-CoV-2. Indicate how to identify indeterminate/equivocal results (if applicable) and how the user should resolve them. Also describe if and when repeat testing may be required.]
[For lateral flow assays: Clearly describe how results are to be interpreted for each of the test lines. You could consider reporting the results in the form of a table as shown below for a test that detects and differentiates between IgG and IgM.]
Table 1. Interpretation of Results
|
C Line |
M Line |
G Line |
Test Result Interpretation |
1 |
not present |
Any |
Any |
Invalid Test. The sample must be retested with another device |
2 |
+ |
- |
- |
Valid Test, Negative for antibodies for SARS-CoV-2 |
3 |
+ |
+ |
- |
Valid Test, IgM positive and IgG negative for antibodies for SARS-CoV-2 |
4 |
+ |
+ |
+ |
Valid Test, IgM and IgG positive for antibodies for SARS-Cov-2 |
5 |
+ |
- |
+ |
Valid Test, IgM negative and IgG positive for antibodies for SARS-CoV-2 |
[Please include a schematic/picture showing the location of the sample well, buffer well, and control and test lines.]
[For assays with qualitative results that are not a lateral flow assay: Clearly describe how results are to be interpreted. If applicable, clearly indicate how to interpret numeric test values as positive or negative for the presence of antibodies against SARS-CoV-2. Indicate how to identify indeterminate/equivocal results (if applicable) and how the user should resolve them. Also describe if and when repeat testing may be required. You could consider reporting the results in the form of a table as shown below.]
Table 2. Interpretation of Results of a qualitative assay (example of an assay with 1 cutoff)
-
Level: Numerical Result in units*
Result
Test Result Interpretation
x < [Cutoff]
Negative
Antibodies for SARS-Cov-2 are not detected.
x > [Cutoff]
Positive
Antibodies for SARS-Cov-2 are detected.
* Numerical results observed with the [test name] are not reported outside of the laboratory since this test is for qualitative use.
Table 3. Interpretation of Results of a qualitative assay (example of an assay with 2 cutoffs and an equivocal zone)
-
Level: Numerical result in units*
Result
Test Result Interpretation
x < [Cutoff 1]
Negative
Antibodies for SARS-Cov-2 are not detected.
[Cutoff 1] < x < [Cutoff 2]
Indeterminate/
Equivocal
Antibodies determination is indeterminate/equivocal with this sample. Test another sample later [specify the timeframe].
x > [Cutoff 2]
Positive
Antibodies for SARS-Cov-2 are detected.
* Numerical results observed with the [test name] are not reported outside of the laboratory since this test is for qualitative use.
For quantitative assays: [Clearly describe how results are to be interpreted.] For these tests, units should be reported as the appropriate CRM units (e.g., BAU/mL, etc.).
Table 4. Interpretation of Results of a quantitative assay (example of a quantitative assay that detects and quantifies antibodies)
-
Level: Numerical result in the CRM units
Result
Test Result Interpretation
x < [Cutoff]
Negative
Antibodies for SARS-CoV-2 are not detected.
[ULMI]* ≥ x ≥ [Cutoff]
Positive; Numerical result is reported outside of the laboratory
Antibodies for SARS-Cov-2 are detected.
> [ULMI]*
Positive; Report outside of the laboratory indicates that the result is above ULMI**
*Upper limit of measuring interval (ULMI)
**Appropriate validation data should be provided if you intend to extend the measuring interval above the ULMI by diluting subject samples.
PRODUCT MANUFACTURING
The [test name] has been validated using only the components referenced in this request and will not be changed after authorization without prior concurrence from the FDA.
The [test name] was developed using [briefly describe the capture antigens, including amino acid sequences, and antibodies used in the test, how they were designed and purified (e.g., are monoclonal antibodies used, are they manufactured in house or purchased commercially, what species they derive from, what epitope is targeted by the antibodies used in the assay, and if commercial products, whether there is a certificate of analysis, etc.).]
Overview of Manufacturing and Distribution
The product will be manufactured at [test developer’s name and FDA registration number (if applicable)] by [test developer’s name] personnel consistent with practices for the production of [types of devices] based on [type of quality system (e.g., 21 CFR 820 or ISO13485)]. Material manufactured by [test developer’s name] may be bottled and kitted by [packager name] manufacturing facility.
The current manufacturing capabilities include the ability to manufacture approximately [please insert the approximate number of tests/kits that can currently be manufactured per week at the manufacturing facility] products per week for distribution in the United States, however, in the event of a surge in demand this could be increased to [please insert the approximate maximum number of tests/kits that could potentially be manufactured per week at the manufacturing facility if there was a surge in demand] products per week within a [please specify in weeks/months the expected timeframe required to increase product production, if conditions warrant] timeframe.
Under an EUA, certain sections of the 21 CFR Part 820 Quality System Regulation (QSR) requirements may be waived for an authorized product during the duration of the EUA, but FDA recommends that test developers follow comparable practices as much as possible, even if such requirements are waived. Please see recent letters of authorization for examples of which QSR requirements have been required.
[Please specify any instruments or other components of your test which are labeled as research use only (RUO) or are otherwise not labeled with the statement “For In Vitro Diagnostic Use” or a symbol found in a standard to the same effect.]
For distributed tests (i.e., tests intended to be performed in more than one laboratory location), that use an RUO instrument, please provide the following information, as applicable:
FOR AN RUO INSTRUMENT WHERE THE EUA REQUESTER IS NOT THE MANUFACTURER OF THE INSTRUMENT:
Please include in the instructions for use found in your test’s labeling, appropriate procedures, including acceptance criteria, that laboratory customers should follow to qualify the performance of the RUO instrument prior to use with your test.
These procedures could include wet testing of quantitated test material with your test, or confirmation that key specifications of the instruments that are applicable to your test are within an appropriate range. The quantitated test material could either be positive control material included with your kit or commercially available positive control material. If commercially available material is not labeled with the statement “For In Vitro Diagnostic Use” or a symbol found in a standard to the same effect, then you should qualify lots of this material in-house and have a mechanism to notify laboratory customers which lots are appropriate to use for qualification (i.e., posting on a website). For the qualification protocol, you should include a recommendation to test multiple dilutions of test material with your test, with, at minimum, 3 replicates per dilution. There should be at least one dilution near the assay cutoff (i.e., two to three times the cutoff) of your test. The protocol should outline the acceptance criteria for each dilution tested.
[Please also provide the following labeling documentation with your request:
A "For Emergency Use Authorization only" label that users can affix to the instrument after the instrument has been qualified. This can be provided as an Appendix in the assay instructions for use.
Please ensure that your test’s labeling either reproduces the parts of the instrument operating manual that are relevant to run your test or references the relevant sections of the manual.]
FOR AN RUO INSTRUMENT WHERE THE EUA REQUESTER IS THE MANUFACTURER OF THE INSTRUMENT:
[Please either provide the qualification protocol as described above or the following information to demonstrate your instrument meets the minimum quality system requirements for authorization.
The ISO 13485 certificate for the site where your instrument is manufactured.
A document mapping out the parts of your quality system that fulfill the following:
Subpart H (Acceptance Activities, 21 CFR 820.80 and 21 CFR 820.86),
Subpart I (Nonconforming Product, 21 CFR 820.90), and
Subpart O (Statistical Techniques, 21CFR 820.250).
Please provide the following labeling documentation with your request:
A “For Emergency Use Authorization only” label that the users can affix to the RUO instrument after it has been qualified. This can be provided as an Appendix in the assay instructions for use.
The instrument operating manual. Please note that the manual should not include any unapproved, uncleared, or unauthorized uses.
An instrument manual addendum that will be distributed along with your EUA test kit. The addendum may have the following format:]
Instrument Operation Manual Addendum:
For emergency use authorization only with the [test name].
The [test name] is authorized for use under the US Food and Drug Administration (FDA) Emergency Use Authorization (EUA) with the [name of instruments] for the [presumptive] qualitative detection of antibodies to SARS-CoV-2 [intended use of test]. Refer to the [test name] instructions for use for additional information [provide hyperlink].
This instrument operation manual addendum applies to the instruments listed in Table 5 that are authorized for use with the [test name].
Table 5: Instruments Authorized for Emergency Use Only with the [test name]
Catalog Number |
Product Name |
|
|
|
|
|
|
|
|
Warnings:
This product has not been FDA cleared or approved; the product has been authorized by FDA as part of [test name] under an EUA for emergency use only by authorized laboratories certified under the Clinical Laboratory Improvement Amendments of 1988 (CLIA), 42 U.S.C 263a.
This product has been authorized only for detecting antibodies to SARS-CoV-2, not for any other viruses or pathogens.
The emergency use of this product is only authorized for the duration of the declaration that circumstances exist justifying the authorization of emergency use of in vitro diagnostics for detection and/or diagnosis of COVID-19 under Section 564(b)(1) of the Federal Food, Drug, and Cosmetic Act, 21 U.S.C. § 360bbb-3(b)(1), unless the declaration is terminated or authorization is revoked sooner.
Components & Other Materials/Information Included with the Test:
Components manufactured by [test developer’s name and FDA registration number (if applicable)] and other materials/information that are supplied with the test include:
[List all components and other materials/information included with your test, including a description of the reagents, volumes, concentrations, quantities, buffer components, etc.]
Table: Example Kit Components & Other Materials/Information
Kit Components & Other Materials/Information |
Main Reagents composition/ Matrix |
Concentration/ Quantity/Volume |
Manufacturer |
Test Cassette with test strip |
|
|
|
Negative control |
|
|
|
Positive control |
|
|
|
Calibrators |
|
|
|
Sample buffer (bottle) |
|
|
|
Transfer pipette |
|
|
|
Lancet (for fingerstick only) |
|
|
|
Instructions for Use leaflet |
|
|
|
Packing materials |
|
|
|
Other Materials/Information, as applicable |
|
|
|
Components and Other Materials/Information Required but Not Included with the Test:
[List all components and other materials/information (e.g., timer, analyzer/reader, reagents) not included with the test that must be supplied by the user to perform the test, with specific supplier names and catalog numbers or other identifiers for obtaining the components. Please include here all specific consumables that were validated for use with your device, that are not interchangeable with other products and that are needed to guarantee device performance, as established in the EUA validation studies listed in Section J below.]
Software Validation:
If you are introducing a system onto the market that has not been previously reviewed by FDA, we recommend providing evidence that the software has been validated to ensure that:
The inputs and outputs of the software are appropriate to fulfill the system and assay requirements;
All expected inputs produce the expected outputs for all functions critical for system operation; and
The system will be provided to the customer free of defects, or defects will be known and mitigated.
If this evidence is not available prior to authorization and the software and hardware have been designed and developed in a manner consistent with current GMPs (for additional information, please see the discussion of “Quality System Regulation/Medical Device Good Manufacturing Practices,” on the FDA website10), additional software validation documentation may be incorporated into the conditions of authorization. If changes which impact assay performance or safety and effectiveness of the system are needed to address validation failures post-authorization, an EUA supplement may be required under the conditions of authorization.
Below are examples of tables for providing system specific information and your evidence that specifications have been met (e.g., hazard analysis). Text in the tables is provided as an example only. [Please provide thorough functional descriptions of system software and instrumentation specifications needed to support the intended use of the test, and provide evidence that specifications have been fulfilled.]
System specifications and validation example
Critical specifications: Description of the specification |
Evidence that the design of the system can fulfill the specification. This column should consist of system-level validation data. |
Optical system of each instrument sent to a user has sufficient dynamic range to appropriately differentiate between positive and negative test results |
|
Software displays appropriate result during test run |
|
If reader stores test result, software accurately stores and retrieves test results |
|
System has a defined lifetime where the user can expect the system to maintain performance as stated in the label |
|
Etc. |
|
Hazard analysis examples
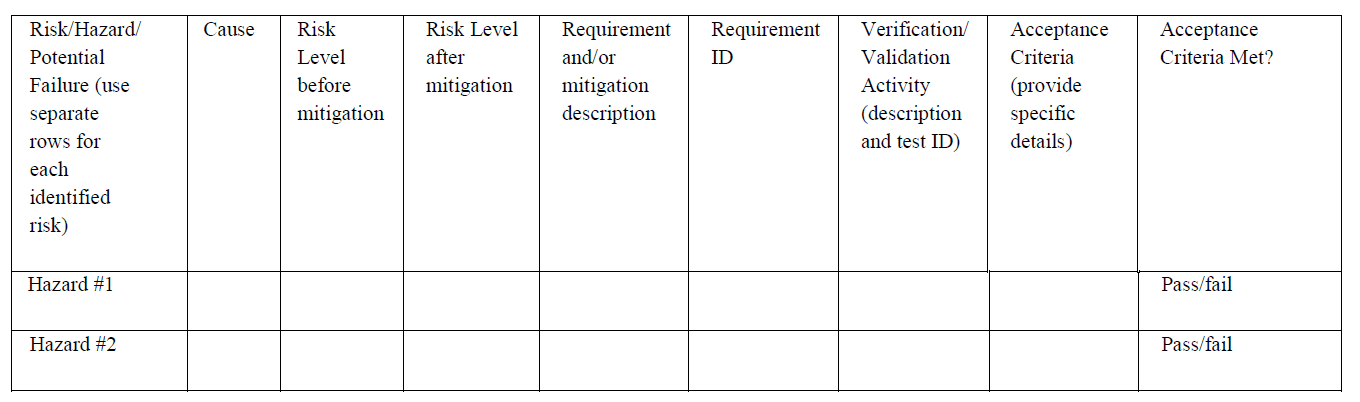
-
ID
Hazard
Adverse Effect
Severity
Potential causes of hazard
Risk mitigation measure
Risk of experiencing the hazard after mitigation
1
Invalid result
Delay in returning test result
Low
User inserts cartridge incorrectly
Labeling noting correct orientation
Low
2
False result
Wrong result returned to user
High
Incorrect alignment of test strip and optics; test strip inserted in the wrong orientation
Mechanical design of reader input slot
Moderate
3
False negative result
Wrong result returned to user
High
User reads test strip too early; incubation time not sufficient
Labeling noting correct incubation time
Moderate
4
False result
Wrong result returned to user
High
Incorrect alignment of test strip and optics; control line misinterpreted
Software interprets data from optical system identifying a valid/invalid control
Moderate
5
False result
Wrong result returned to user
High
Control reaction intensity is misinterpreted
Software interprets data from optical system identifying a valid/invalid control
Moderate
6
False result
Wrong result returned to user
High
Analyte reaction intensity is misinterpreted
Software interprets data from optical system identifying a valid/invalid control
Moderate
If applicable to your test, FDA recommends the following evaluations be performed and documentation kept on file. If not completed by the time of authorization, these evaluations may be required as a condition of authorization.
You should evaluate the cybersecurity of your system to ensure user and patient safety in the intended use environment;11
You should complete validation of all systems and software to ensure that all functions of the system perform as labeled. For more information on system validation please see the following FDA guidance documents and resources:
Basic Safety and Essential Performance:
[If you are introducing a system onto the market which has not been previously reviewed by the FDA, please describe how you addressed basic safety hazards such as electrical hazards (e.g., electrical shock to the operator and/or patient), fire hazards, and mechanical hazards.] We recommend that you consult the general requirements for basic safety, as indicated in International Electrotechnical Commission (IEC) 60601-1 (Medical electrical equipment – Part 1: General requirements for basic safety and essential performance). IEC 60601-1 is a standard that specifies the general requirements for basic safety and essential performance. IEC 60601-1 defines basic safety as freedom from unacceptable risk directly caused by physical hazards when medical electrical equipment is used under normal condition and single fault condition.
Electromagnetic Compatibility (EMC) Testing (if applicable):
We recommend that EMC testing be conducted on any assay that uses a battery or power source. [Please provide FDA with any standards that were followed for EMC testing.] We recommend that you perform EMC testing according to the International Electrotechnical Commission (IEC) 60601-1-2 Edition 4.0:2014. [If you perform EMC testing to a different standard or use alternate methodologies to evaluate EMC, please provide a test plan, test report, acceptance criteria, and risk analysis to support your approach.]
Manufacturing and Testing Capabilities
[Briefly describe current sample throughput testing capacity, the total time required to perform the test (from clinical sample collection to result), and the number of tests that can be performed per day (8-hour shift), excluding controls and calibrators, as applicable. Please provide the number of kits you can manufacture per day/week for distribution in the United States.]
Distribution Plan:
The product will be distributed by [please describe the distribution plan for the product and list all current US distributors.].
Reagent Stability
[Briefly describe the stability test plan for [test name] reagents.] Real time reagent stability studies generally would not need to be completed at the time of EUA issuance, however, the study design should be agreed upon during interactive review and the stability studies started immediately following authorization, if not before. You should consider the following general recommendations when designing your reagent stability study:
For EUAs, you may follow the current FDA recognized Clinical Laboratory Standards Institute (CLSI) EP25, “Evaluation of Stability of In Vitro Diagnostic Reagents: Approved Guideline, when evaluating the suitability of stability study designs. If you are planning to pursue a clearance or approval for your device, we recommend discussing with FDA in more detail your stability design to facilitate potential use of the EUA data in your regular premarket submission.
We recommend testing a known positive clinical sample or sample type contrived from positive samples rather than positive control material to establish reagent stability. You should test low – moderate positive samples that are expected to produce positive results in the serology assay 100% of the time.
If you are using multiple clinical sample types, you should use the most challenging clinical matrix for this study (e.g., whole blood).
You should evaluate at least 5 replicates and if available, 3 different lots of reagents.
You should design your study to provide data for a timeframe that is about 10% longer than the one to be authorized. For example, 18 months should be supported by stability data out to 20 months and 7 days should include stability data out to 8 days.
FDA considers 15-30°C to represent room temperature conditions. Ideally, you should evaluate stability at both 15°C and 30°C; however, for the purposes of the EUA evaluation, 30°C is generally appropriate, as it represents the worst-case scenario.
Unopened Kit Shelf-Life Stability:
You should evaluate real-time kit stability studies with unopened kits stored at the claimed storage temperature for your test.
Please note real-time stability data should be provided to support shelf-life claims at EUA issuance; alternatively, unopened kit shelf-life claims of 9 months at 2-8 °C may be considered at EUA issuance while real-time studies are on-going.
Unopened Kit Shipping Stability: You should evaluate the anticipated handling and shipping times and temperatures expected for unopened kits under different temperature conditions (e.g., summer, winter). The recommended summer profile is storage at 40oC for 8 hours and then 22oC for 4 hours and the recommended winter profile is -10oC for 8 hours and then 18oC for 4 hours.
In-use/Opened Kit Stability: Depending on your device, your stability study design should also support in-use stability of the kit reagents once the kit has been opened, e.g., storage at 2-8°C for 7 days. This includes on board stability once reagents have been placed on the instrument (if applicable).
Inverted stability (if applicable): Study should support stability for kits if stored inverted or in the wrong orientation.
FDA recommendations for analysis of real time stability studies are as follows:
Baseline of the study (t=0 of stability study) should not exceed one month from production;
Clear baselines should be described (e.g., one month from production) for each stability claim under each study;
Claims should be determined based on regression analysis. Any %change (%shift) from time zero (baseline) should be calculated between the target claim and the zero-time as (Ttest-Tbaseline)/ Tbaseline*100 with 95% confidence interval (CI) using the regression equation obtained from plotting the mean values. When formulating your acceptance criteria for evaluating the shift from baseline. you should consider the reproducibility of your device. Generally, the shift at the target claim due to storage should not exceed 10-15%. The target stability is the next to last tested point that was within +/- 10% of time zero; and
Acceptance criteria may differ depending on the reproducibility of your device, the distribution of analyte concentration expected in samples from the intended use population, and the risk of false results to public health.
PERFORMANCE EVALUATION
The following validation studies should be performed to support your EUA request. Please note that, particularly for new technologies, FDA may request additional studies so we can adequately assess the known and potential risks and benefits associated with the candidate test. [For each validation study, you should provide a study protocol that includes a detailed, step-by-step description of how samples were prepared and how testing was conducted. You should also include the study data from each validation study in an Excel-compatible format for all validation studies. The line data should present each replicate with the final candidate serology test result per the tests result interpretation. If the candidate test has a numeric output, you should include the numeric values for each replicate.]
Analytical Sensitivity and Specificity:
Inclusivity (Analytical Reactivity):
Mutations in the SARS-CoV-2 genome have been identified as the virus has spread. A mutation is an individual genetic change in a SARS-CoV-2 virus sequence when compared with a reference sequence such as Wuhan-Hu1 or USA-WA1/2020. A new virus variant of SARS-CoV-2 has one or more mutations that differentiate it from the wild type or predominant virus variants already circulating in the general population. Variants of SARS-CoV-2 are identified by genomic sequences that contain mutation(s) in the RNA genome, which could result in amino acid substitutions, insertions, and/or deletions in viral proteins. Different variants can result in different phenotypes (e.g., a difference in antigenicity, transmissibility, or virulence). Viral mutations and viral variants could result in altered immunogenicity relative to the originally isolated virus, which could impact the performance of serology tests. For example, some studies suggest that identified variants (which may contain multiple mutations) may affect the ability of some antibodies to neutralize the virus in vitro.
Test developers should monitor new and emerging viral mutations and variants, that could impact serology test performance on an ongoing basis. This includes assessing the prevalence of viral mutations in sequence databases (e.g., the GISAID16 database), as mutations observed in these databases at a significant frequency may signify that the mutation is present in an increasing proportion of infected individuals in the U.S. FDA currently considers a significant frequency to be greater than 5% (when considering at least 2000 sequences over a recent period of time, such as the past week, month, or quarter). Monitoring should also include identifying if there are multiple credible reports indicating that a given viral variant (which may have one or more mutations) has the potential to increase virulence, increase transmission, or otherwise increase the public health risk. FDA recommends monitoring on at least a monthly basis in light of the rate of occurrence of mutations and variants and the importance of assessing their impact.
For any viral mutations and variants that are identified as prevalent and/or clinically significant as described above, you should assess whether the resulting predicted amino acid change(s) in the viral proteins are critical to your test design. If the mutations are found to be critical to your test design, mutations and variants should be evaluated using clinical (or contrived, as available and as appropriate) samples to assess the impact of the mutation or variant on your test’s performance.
The aggregate impact of the mutations should not reduce the clinical performance of the test by 5% or more, or decrease the clinical performance point estimates for the test below the clinical performance recommendations described in Section J.3.d.17
Please see the FDA guidance document “Policy for Evaluating Impact of Viral Mutations on COVID-19 Tests” for additional discussion regarding monitoring the impact of genetic variants on serology tests. 18
FDA also has ongoing monitoring efforts and may identify a viral mutation or variant as clinically significant for which testing with clinical (or contrived, as available and as appropriate) samples would be recommended to assess the impact of the mutation or variant on the performance of your test.
[Please provide your plan for monitoring for new and emerging SARS-CoV-2 viral mutations and variants on an ongoing basis and for assessing the impact of mutations and variants that have been identified as prevalent and/or clinically significant on the performance of your assay over time.]
[For mutations and variants that have been identified as prevalent and/or clinically significant as part of ongoing monitoring at the time of your EUA request, please provide information on the potential impact of the mutation(s) and variants on your test’s performance or explain how the risk associated with the unknown performance of your device in samples from individuals with the variant(s) can be adequately mitigated.]
Cross-Reactivity (Analytical Specificity):
If a large number of known negative samples (e.g., ≥75 unique samples collected in the US prior to December 2019) are tested from a population with a high prevalence of vaccination against, and/or infection with, the following viruses, and specificity >95% is observed, cross-reactivity testing for the following viruses would not be expected at this time:
anti-influenza A (IgG and IgM) |
anti-influenza B (IgG and IgM) |
anti-HCV (IgG and IgM) |
anti-HBV (IgG and IgM) |
anti-Haemophilus influenzae (IgG and IgM) |
anti-229E (alpha coronavirus) |
anti-NL63 (alpha coronavirus) |
anti-OC43 (beta coronavirus) |
anti-HKU1 (beta coronavirus) |
ANA |
anti-respiratory syncytial virus (IgG and IgM) |
anti-HIV |
[If a large number of known negatives are evaluated (with at least 95% specificity observed) rather than specific samples with the cross-reactants above, please include information adequate to demonstrate that the samples tested were from a population with a high prevalence of vaccination against and/or infection with, the above viruses including a description of the samples tested, e.g., geographical location and any other information you have available regarding how the samples were collected and sourced.]
[If a large number of known negative samples are not evaluated, describe the cross-reactivity testing performed to evaluate the cross-reactants in the table above. Please include in your description the number of samples tested and how samples were prepared.]
If testing of the cross-reactants is needed to demonstrate cross-reactivity of the test, a minimum of 5 individual samples for each disease/infectious agent/antibody class listed above should be evaluated.
If natural clinical samples are used, it is important to assess cross reactivity using sera from individuals with the underlying diseases in the acute or convalescent stages of infection or individuals vaccinated for specific infectious diseases, in order to obtain high levels of IgM or IgG for the underlying condition. If spiked samples with the IgM or IgG antibodies for the underlying conditions are prepared for this study, it is important to confirm that “negative samples” are SARS-CoV-2 IgM and IgG seronegative with the candidate test prior to spiking. Additionally, commercially available IgM or IgG antibodies for the underlying conditions panels may be appropriate if collected prior to the COVID-19 pandemic, to ensure the panels are SARS-CoV-2 antibody-negative.
We recommend you present your results in the following table and calculate agreement between the candidate test result and the expected result.
Table Cross-Reactivity: [test name] example table for wet tested organisms below:
-
Virus/Bacteria/Parasite Antibody positive
Source/ Sample type
Results*
*If applicable, please include the signal output for your test’s technology.
[If your test exhibits significant cross-reactivity that would produce false positive results for any virus evaluated, please describe a plan that would adequately mitigate the risk.]
Class Specificity (if applicable)
If your test is intended for the detection of total antibody with no differentiation between different immunoglobulins, then this study does not apply. [In this case, please indicate that this study is not applicable.]
[If your test is intended to differentiate between different immunoglobulins, describe the approach used to evaluate class specificity.]
Approaches to evaluate class specificity depend on the assay format. If you have used well-characterized anti-IgG and anti-IgM reagents in your test, class specificity testing may not be needed. In this case, FDA recommends describing how the reagents were characterized and how such characterization supports class specificity.
[If class specificity testing is needed for your test, please describe the study, or studies, performed to demonstrate that the assay accurately detects each antibody class (e.g., IgG and IgM). This should include a description of the studies performed to evaluate the potential for human IgM to cross react and therefore produce false positive results for IgG, and the reverse, and the potential for IgM to compete with IgG and produce false negative results. Please indicate the number of samples, and the number of replicates per sample, tested. Evaluating at least 5 samples positive for both antibody classes (IgM positive while also IgG positive), in duplicate, may be appropriate if an IgG-only and IgM-only positive control are included in the study as indicated in the example table below. Commercially available positive controls for this study are appropriate (e.g., monoclonal, recombinant antibodies). Please provide the protocol and results, including line data, from any class specificity testing.]
One recommended approach includes treating the samples with dithiothreitol (DTT) where the final IgG result will remain unaffected, and the final IgM signal will decrease or be negative. A positive control should also be included that confirms DTT activity.
100% agreement with the expected result would establish antibody class specificity.
If a DTT Treatment approach is followed, below is an example table for IgM and IgG:
Clinical Agreement StudySample
ID
Replicates
Result
NO DTT Treatment
(IgM/IgG)
Result
DTT Treatment
(IgM/IgG)
Expected result with DTT treatment
(IgM/IgG)
Result Agreement
1
1
+/+
-/+
-/+
yes
2
+/+
-/+
-/+
yes
2
1
+/+
-/+
-/+
yes
2
+/+
-/+
-/+
yes
3
1
+/+
-/+
-/+
yes
2
+/+
-/+
-/+
yes
4
1
+/+
-/+
-/+
yes
2
+/+
-/+
-/+
yes
5
1
+/+
-/+
-/+
yes
2
+/+
-/+
-/+
yes
IgG only positive control
1
-/+
-/+
-/+
yes
2
-/+
-/+
-/+
yes
IgM only positive control
1
+/-
-/-
-/-
yes
2
+/-
-/-
-/-
yes
[Please describe the clinical study used to evaluate the clinical performance of the test. Please note that the recommendations for the clinical evaluation depend on access to COVID-19 disease clinical samples at the time of the studies and the nature of the emergency.]
Initial clinical agreement studies typically evaluate all matrices that developers intend to include in their EUA request.
[Please specify how the samples were generated, collected, and sourced. Please also specify if the samples were fully prospective or a mix of prospective, retrospective and/or contrived. Please specify inclusion/exclusion criteria, collection and testing sites, number of samples collected and tested, and number of operators performing the testing.]
Comparator
At this time, the comparator method used to establish a subject’s prior infection with SARS-CoV-2 is a reverse transcription polymerase chain reaction (RT-PCR) based test. Results from the FDA authorized RT-PCR comparator test are obtained using samples that have been validated for use with that test (for example, nasopharyngeal swabs, nasal swabs, etc.). Samples tested to confirm SARS-CoV-2 infection by RT-PCR should be collected prior to samples tested on the candidate test. Individuals should be previously confirmed as infected with SARS-CoV-2 by an FDA authorized RT-PCR comparator test using nasal swab samples, and serum, plasma, venous whole blood, and/or fingerstick whole blood should be collected from the same individuals at later dates for serology testing with the candidate test.
[Please identify the FDA authorized RT-PCR comparator test that was used. The FDA authorized RT-PCR comparator test used for each sample should be documented; it is generally not appropriate to rely on self-reported RT-PCR results from study subjects.] We recommend using only a high sensitivity FDA authorized RT-PCR comparator test which uses a chemical lysis step followed by solid phase extraction of nucleic acid (e.g., silica bead extraction) as the comparator test; for example, an FDA authorized RT-PCR comparator test that has established high sensitivity with an internationally recognized standard or FDA SARS-CoV-2 Reference Panel.19 Please check with FDA if unsure.
Clinical agreement study population
Ideally, performance characteristics are established in a clinical study with prospective samples collected in the United States to reflect the intended use population of the device. If a prospective study is not feasible, an appropriate approach would be to test archived samples from individuals that have been previously confirmed infected by an FDA-authorized SARS-CoV-2 RT-PCR comparator test, accompanied by basic information such as the population from which the sample was drawn, sample collection date, date of onset of symptoms, FDA authorized RT-PCR comparator test used to confirm individuals as SARS-CoV-2 infected or not infected (see above), and FDA authorized RT-PCR comparator test collection/testing date. To avoid introducing bias into the study, samples should not be pre-selected with an alternate serology test prior to testing with an FDA-authorized SARS-CoV-2 RT-PCR comparator test and the candidate test.
Clinical agreement sample size
Clinical agreement data should be provided using at least 30 unique samples collected from 30 RT-PCR positive individuals for each immunoglobulin claimed and 75 unique samples collected from 75 RT-PCR negative subjects tested for SARS-CoV-2, or 75 unique samples collected prior to December 2019.
For all tests, blinding and randomization should be incorporated into the study design.
Clinical agreement data analysis and acceptance criteria
For tests that detect and differentiate IgM and IgG, clinical agreement data should demonstrate a minimum of the following:
Overall (i.e., combined IgM/IgG) positive percent agreement (PPA) of 90%,
PPA for IgM of 70%,
PPA for IgG of 90%, and
Overall (i.e., combined IgM/IgG) negative percent agreement (NPA) of 95%.
For tests that detect either total antibodies, only IgG, or only IgM, clinical agreement data should demonstrate a minimum of the following:
PPA of 90% and
NPA of 95%
If a larger number of unique samples are evaluated (i.e., more than 30 samples collected from unique RT-PCR positive individuals and 75 unique negative samples), the following point estimates and confidence intervals may be appropriate:
NPA not lower than 93% with a lower bound of the 95% confidence interval greater than 87.8%.
For tests that detect either total, only IgG, or only IgM, PPA not lower than 87% with a lower bound of the 95% confidence interval greater than 74.4%.
For tests that report IgG and IgM, PPA for overall (i.e., combined IgM/IgG) not lower than 87% with a lower bound of the 95% confidence interval greater than 74.4%, PPA for IgM not lower than 67% with a lower bound of the 95% confidence interval greater than 52.1%, and PPA for IgG not lower than 87% with a lower bound of the 95% confidence interval greater than 74.4%.
If, in addition to claiming serum, plasma, and/or venous whole blood where clinical agreement is demonstrated as described above, a claim for fingerstick is desired, a minimum of 30 fingerstick samples collected from RT-PCR positive individuals and 30 fingerstick samples collected from RT-PCR negative individuals should be evaluated to demonstrate clinical performance in fingerstick samples. If a claim for point-of-care (POC) testing is desired, please see the section specific to POC below.
[Please clearly describe the data analysis methods used and provide the results from the study, including line data. Positive and negative percent agreement between the candidate test and the FDA authorized RT-PCR comparator test results should be calculated separately for each claimed matrix. Stratification of the data by the number of days since symptom onset should be provided. If the time between generation of the candidate test result and symptom onset is not known, stratifying by days since RT-PCR sample collection may be appropriate. FDA recommends providing the results using a tabular format, examples of which are provided below.]
Clinical performance data stratified by time from symptom onset, using the time periods below, should, at a minimum, meet the performance metrics specified above for PPA/sensitivity for the >15 days from symptom onset time period (or 15-30 days from symptom onset time period for IgM only tests).
If 30 or more samples are collected ≥15 days from symptom onset (or 15-30 days from symptom onset time period for IgM only tests), the performance of those samples should meet the clinical performance estimates above; OR
If less than 30 samples are collected ≥15 days from symptom onset (or 15-30 days from symptom onset time period for IgM only tests), at least 10 samples should be collected ≥15 days from symptom onset and meet the clinical performance estimates above and
The clinical performance estimates for the 8-14 day time period and the ≥15 day time period both meet the performance metrics above and the total number of samples collected ≥8 days (i.e., samples in the 8-14 and ≥15 day time periods) is at least 30 samples; OR
The clinical performance estimates for the 0-7 day, 8-14 day, and the ≥15 day time periods all meet the performance metrics above and the total number of samples collected in all three time periods is at least 30 samples.
Example performance tables
For Total Antibody Tests:
The following table is an example of how you may present the total antibody PPA by time of sampling from symptom onset (in subjects confirmed to have been previously infected with SARS-CoV-2 by a prior positive RT-PCR result):
|
Candidate Test Results |
|||
Days from Symptom Onset |
Number of Subjects Tested |
Total Antibody Positive results |
Total Antibody PPA |
95% CI |
0-7 days |
[A] |
[a] |
[a/A (%)] |
|
8-14 days |
[B] |
[b] |
[b/B (%)] |
|
≥15 days |
[C] |
[c] |
[c/C (%)] |
|
The following table is an example of how you may present the NPA using RT-PCR negative samples or samples assumed to be negative since they were collected prior to December 2019:
-
Candidate Test Results
Number of Subjects Tested
Negative Results
Overall NPA
(95% CI)
[D]
[d]
[d/D (%)]
Tests that detect and distinguish IgG and IgM antibodies:
If you claim that your test can differentiate between IgG and IgM, PPA and NPA for IgG and IgM should also be calculated separately, as in the example below.
The following tables are examples of how you may present the IgG or IgM PPA by time of sampling from symptoms onset (in subjects confirmed to have been previously infected with SARS-CoV-2 by a prior positive RT-PCR result):
-
Candidate Test Results
Days from Symptom Onset
Number of Subjects Tested
IgG Positive results
IgG PPA
95% CI
0-7 days
[A]
[a]
[a/A (%)]
8-14 days
[B]
[b]
[b/B (%)]
≥15 days
[C]
[c]
[c/C (%)]
-
Candidate Test Results
Days from Symptom Onset
Number of Subjects Tested
IgM Positive results
IgM PPA
95% CI
0-7 days
[A]
[a]
[a/A (%)]
8-14 days
[B]
[b]
[b/B (%)]
≥15 days
[C]
[c]
[c/C (%)]
15-30 days
[D]
[d]
[d/D (%)]
-
Candidate Test Results
Days from Symptom Onset
Number of Subjects Tested
IgG/IgM Combined Antibody Positive results
IgG/IgM Combined Antibody PPA
95% CI
0-7 days
[A]
[a]
[a/A (%)]
8-14 days
[B]
[b]
[b/B (%)]
≥15 days
[C]
[c]
[c/C (%)]
The following table is an example of how you may present the NPA using RT-PCR negative samples or samples assumed to be negative since they were collected prior to December 2019:
-
Candidate Test Results
Number of Subjects Tested
IgM/IgG Negative Results
NPA
(95% CI)
[D]
[d]
[d/D (%)]
IgG only antibody tests:
If you claim that your test can detect IgG only, PPA and NPA for IgG should be calculated, as in the example below.
The following tables are examples of how you may present the IgG PPA by time of sampling from symptom onset (in subjects confirmed to have been previously infected with SARS-CoV-2 by a prior positive PCR result):
-
Candidate Test Results
Days from Symptom Onset
Number of Subjects Tested
IgG Positive results
IgG PPA
95% CI
0-7 days
[A]
[a]
[a/A (%)]
8-14 days
[B]
[b]
[b/B (%)]
≥15 days
[C]
[c]
[c/C (%)]
The following table is an example of how you may present the NPA using PCR negative samples or samples assumed to be negative since they were collected prior to December 2019:
-
Candidate Test Results
Number of Subjects Tested
IgG Negative Results
IgG NPA
(95% CI)
[D]
[d]
[d/D (%)]
IgM only antibody tests:
If you claim that your test can detect IgM only, PPA and NPA for IgM should be calculated, as in the example below.
The following tables are examples of how you may present the IgM PPA by time of sampling from symptom onset (in subjects confirmed to have been previously infected with SARS-CoV-2 by a prior positive RT-PCR result):
-
Candidate Test Results
Days from Symptom Onset
Number of Subjects Tested
IgM Positive results
IgM PPA
95% CI
0-7 days
[A]
[a]
[a/A (%)]
8-14 days
[B]
[b]
[b/B (%)]
15-30 days*
[C]
[c]
[c/C (%)]
*Note that the days from symptom onset for IgM only antibody tests differ from the days from symptom onset used for IgG, IgG/IgM, and total antibody tests.
The following table is an example of how you may present the NPA using RT-PCR negative samples or samples assumed to be negative since they were collected prior to December 2019:
-
Candidate Test Results
Number of Subjects Tested
IgM Negative Results
IgM NPA
(95% CI)
[D]
[d]
[d/D (%)]
Matrix Equivalency
[Please describe the protocol for and provide the results from any matrix equivalency studies performed to support the performance of the assay in the sample types used (serum, Ethylenediaminetetraacetic acid (EDTA) plasma, venipuncture whole blood, different anticoagulants, etc.) that were not evaluated in the initial clinical agreement study. For assays with a numerical value result, FDA recommends providing regression analysis (e.g., Deming or Passing-Bablok regression) comparing the results obtained with the comparator sample type and the other sample types.]
Please note: Fingerstick whole blood is not considered to be the same sample type as venipuncture whole blood. Test performance with fingerstick whole blood should be evaluated by clinical agreement against RT-PCR and not in a matrix equivalency study (please see Section J.3 above).
Matrix equivalency studies are performed to evaluate sample matrices for which clinical agreement is not initially assessed. In these studies, the matrix in which the clinical study(ies) is/are conducted is the comparator matrix/sample type and each matrix set (whole blood, plasma, serum) comes from the same donor (i.e., paired/matched samples).
Typically, negative, low positive (e.g., for lateral flow tests, samples expected to generate a faint test line, for tests that have an underlying signal value, two to three times the assay cut-off), and moderate positive (e.g., for lateral flow tests, strong test line and for tests that have an underlying signal value, five times the assay cut-off) are evaluated. [Please include a detailed description of how the samples were prepared and the method used to establish the level of SARS-CoV-2 antibodies present in these samples prior to conducting the matrix equivalency study.] Five samples, run in duplicate for each concentration, for a total of 30 results per matrix (assuming 3 concentrations were evaluated) should be evaluated. To allow for comparison, negative samples for each sample type/matrix are spiked with the same amount of analyte (SARS-CoV-2 antibodies). Positive natural clinical samples should be used to spike negative clinical samples since recombinant antibody may not represent the diversity of antibodies found in native samples. Confirming samples are antibody seronegative with the candidate test before spiking with SARS-CoV-2 antibodies is important. When spiking negative matrix with SARS-CoV-2 antibody-positive samples, in order to ensure matrix integrity, no more than 15% of the total sample volume should be from the spiking material.
For all tests, blinding and randomization are important considerations for the study design.
For these types of studies, typically, each sample is assayed with the candidate test, and the results obtained for the comparator matrix are compared to the results obtained for each additional matrix under evaluation for each subject. PPA and NPA for each matrix with respect to the comparator matrix are calculated. At least 95% PPA and 95% NPA for each matrix comparison (e.g., serum versus EDTA plasma, etc.) may be appropriate to demonstrate that performance between the matrices can be considered equivalent.
Studies to support Point of Care (POC) Use, as applicable:
[If the device is intended for POC testing, please provide a detailed study description and data to demonstrate that non-laboratory healthcare providers can perform the test accurately in the intended use environment (i.e., a non-laboratorian healthcare provider clinical agreement study). Please also provide data to demonstrate robust use of your device for POC testing (e.g., as applicable, studies to demonstrate the impact of adding different volumes of sample, different volumes of reagents, incorrect order of sample or reagent application). Data for each sample evaluated (i.e., line data) should be provided. If erroneous results are observed during studies evaluating the robustness of the device, adequate mitigation(s) should be provided.]
To demonstrate that non-laboratory healthcare providers can perform the test accurately, the following should be considered in the study design (this clinical study design does not apply to testing sites where NO healthcare providers are present):
Performing the testing at one or two point of care sites (e.g., sites operating under a CLIA Certificate of Waiver) sites in the United States (U.S.) to assure that the operators are representative of operators in the U.S. These may be inpatient or external sites.
Testing performed using natural, not contrived, samples.
Testing performed by multiple operators.
If the study to support the POC indication will be the only clinical agreement study to be performed for the EUA request, clinical agreement data should include a minimum of 30 positive fingerstick samples from subjects previously confirmed to be SARS-CoV-2 positive by RT-PCR (with information on day of symptom onset and days post RT-PCR testing) and a minimum of 75 SARS-CoV-2 RT-PCR negative fingerstick samples. If the study is in addition to separate laboratory-based clinical agreement studies, a minimum of 30 positive fingerstick samples and 30 negative fingerstick samples may be sufficient.
Testing should be performed with quick reference instructions (QRI) only; supplemental on-line materials/videos are encouraged but should not be used during the study.
For positive and negative samples, the FDA authorized RT-PCR test and result should be provided.
For negative samples:
If you intend only to claim fingerstick whole blood as a sample type for use at the POC, the RT-PCR status of each sample should be provided and the serology sample should be collected within one week of the RT-PCR collected sample.
If you intend to claim additional sample types and are able to demonstrate adequate clinical performance in those sample types (see Sections J.3. and J.4. above), submitting the RT-PCR status of each sample is encouraged but may not be necessary if the subject is determined to be COVID-19 negative based on a clinical assessment and this information is provided.
FDA acknowledges that in prospective clinical agreement studies intended to support POC claims, it is not possible to test presumptive negative samples collected prior to December 2019 and many individuals may have been exposed to SARS-CoV-2 virus. To address samples collected from RT-PCR negative individuals that may be truly positive for antibodies to SARS-CoV-2 (e.g., from prior exposure, etc.), the following additional testing data and analysis may be considered. This approach provides mitigations to reduce the bias introduced when additional testing results may impact the study population included in the analysis and the resulting performance calculations.
Performance characteristics should be established in a clinical study with unique prospective samples to reflect the intended use population of the device. The negative RT-PCR result of each subject should be documented, and the serology sample should be collected within 1 week of the date that the sample is collected for the RT-PCR test. [You should record and provide in your EUA request basic information such as how the samples were generated, collected, and sourced, the population from which the sample was drawn, inclusion/exclusion criteria, serology sample collection date, collection and testing sites, number of samples collected and tested, number of operators performing the serology testing, the FDA authorized RT-PCR test used, and the RT-PCR collection/testing date.]
For each SARS-CoV-2 RT-PCR negative individual, in addition to the fingerstick sample collected for testing with the candidate test , a paired venous blood draw sample (i.e., serum or plasma. as appropriate) should be collected for testing with additional FDA-authorized serology tests. For all tests, blinding and randomization are important considerations for the study design.
The additional serology testing should be conducted for all RT-PCR negative study subjects according to the instructions for use of the additional serology tests. For this analysis, it is generally not appropriate to test only samples with candidate test results that are discrepant from the expected results.
The venous samples should be tested by at least two different FDA-authorized serology tests that meet or exceed the performance and validation criteria described in the table below. If the results of these two tests are different, a third test should be performed. To avoid bias in the study, the candidate test that is the subject of the EUA request should not be used as one of the additional serology tests in this analysis.
The additional serology tests should be independent from one another (e.g., it is generally not appropriate to use the same test run on different instrument platforms) to provide greater support for specificity in this analysis. Please check with FDA if unsure about the FDA-authorized tests selected for this purpose.
If results are positive for SARS-CoV-2 antibodies by at least two (out of a maximum of three) of the additional FDA-authorized tests that meet the criteria outlined in the table below, the paired sample results (i.e., results from the respective samples tested with the candidate test) may be considered for exclusion from the final NPA calculations for the candidate test compared to RT-PCR status.
You should present NPA calculations separately, in two ways:
NPA calculations for all study samples in comparison to RT-PCR status (i.e., all comers in the intended use population without exclusions), and
NPA calculations after excluding samples that are positive by at least two of the additional FDA-authorized serology tests that meet the criteria outlined in the table below.
Performance should be demonstrated for a minimum of 75 unique samples from RT-PCR negative subjects, with a minimum NPA of 95% after sample exclusion.
[You should provide complete study line data in an Excel-compatible format, including a sheet with descriptions of columns and a narrative description of all study exclusions.]
Recommended Criteria for Selecting Additional Serology FDA-Authorized Tests
-
Laboratory-based, FDA-authorized serology testsa
NPA performance has been validated with a minimum sample size of n ≥ 1000, not counting samples collected from blood donorsb
NPA performance ≥ 99%
PPA performance ≥ 95% for at least 30 unique samples collected ≥ 15 days from symptom onset
Is not the candidate test
Tests are independent from one another
a Lateral flow tests should not be used for this purpose.
b Blood donors consist of a population selected to exclude conditions/infections found in the larger intended use population, and therefore are not recommended for use in the proposed analysis. Additional validation may be recommended to support test selection if the minimum NPA criteria cannot be met without including the results of blood donor sample testing.
At least 5-6 different operators should be either observed, complete a questionnaire after completing the test, or provide unstructured feedback after completing the test specifically regarding the test procedure and any concerns with the instructions.
Please refer to the clinical agreement section above for the performance that should be demonstrated by the data from the non-laboratorian healthcare provider clinical agreement study.
To demonstrate robust use of the device for POC testing, the pre-defined study protocol should include the objective of the study, detailed test procedures, materials used, a list of the samples tested (which should be at least five samples per test result outcome (e.g., negative, low positive)), and data analysis methods. Robustness studies for lateral flow devices should be designed to evaluate perturbations in reading time, sample volume, sample diluent volume, temperature and humidity, light, and any unique test characteristics as determined by a risk analysis. For example, if an upright device, evaluating the impact of performing the test in a non-upright position. For each test, samples are typically prepared in whole blood and include at least five negative and five low positive samples. The low positive samples should be prepared at a concentration expected to produce a faint test line result 100% of the time. The method of preparation should be documented in the protocol. Positive natural clinical samples should be used to spike negative clinical samples since recombinant antibody may not represent the diversity of antibodies found in native samples. All results, including invalids and incorrect results, should be documented and reported in the data submitted in the EUA request.
For devices intended for use with fingerstick samples, robustness studies may be conducted using venous whole blood. The following should be considered in the study design for each robustness test.
Reading Time:
You should evaluate test results at reading times fourfold below and three-fold above the recommended reading time. For example, for a test where the recommended read time is 20 minutes, performance of the test should be evaluated at read times of 5, 10, 15, 20, 30, and 60 minutes. If incorrect results are observed, the sponsor should propose adequate mitigations for how the incorrect timing can be addressed.
Sample Volume:
You should evaluate test results at sample volumes two times below and two times above the recommended sample volume. For example, for a test where the recommended sample volume is 10 μL, performance of the test should be evaluated at sample volumes of 5, 10, and 20 μL. If incorrect results are observed at either 5 or 20 µL, additional testing at 7.5 and/or 15 µL may be recommended. The diluent/buffer amount should be that specified in the instructions for use.
Sample Diluent Volume:
You should evaluate test results at diluent/buffer volumes at two times below and two times above the recommended diluent/buffer volume. For example, for a test where the recommended buffer/diluent volume is 2 drops, sample diluent volume testing would be performed to evaluate at least sample diluent volumes of 1, 2, 3, 4 drops. The sample volume added should be that specified in the instructions for use.
Temperature and Humidity:
You should evaluate test results at temperature and humidity extremes that are likely to occur in the United States. We recommend evaluating at 40°C and 95% relative humidity (RH) to mimic a hot and humid climate.
Light:
You should evaluate test results in different lighting conditions that would be expected during POC use of the device, including fluorescent, incandescent, and natural lighting mimicking the outside environment.
Disturbance during analysis:
You should evaluate the effect on expected test results of moving the device while the test is running, such as dropping/moving the test, unplugging the test, receiving a phone call while the mobile app is running, etc.
Unique test characteristics:
You should evaluate unique device characteristics, as determined by a robust risk analysis. For example, if the device is intended to be run upright, we recommend evaluating test results if the device is used horizontally. If the device is to be used with an instrument, we recommend evaluating environmental stability of the electronics.
Studies to support the measuring interval of a quantitative test (if applicable):
Limit of Blank (LoB):
LoB is the highest measurement result that is likely to be observed (with stated probability, typically 95%) for a blank sample (sample with no analyte of interest). LoB should be determined according to the recommendations in CLSI EP17, “Evaluation of Detection Capability for Clinical Laboratory Measurement Procedures; Approved Guideline- Second Edition,” using at least two reagent lots (including two calibrator lots) and performing measurements on at least three days. Five blank samples (known negatives: clinical samples collected prior to December 2019) should be tested in duplicate for each lot and day. This study design will yield 30 replicates of blank samples per lot. The 95th percentile of 30 blank replicates is the LoB estimate for each lot. The LoB of the assay is the maximum value of the individually determined LoBs for each lot.
Limit of Detection (LoD):
LoD is the lowest concentration of analyte that the test can consistently detect with stated probability (usually, 95% of the time). LoD should be determined according to the recommendations in CLSI EP17, using at least two reagent lots (including two calibrator lots) and performing measurements on at least three days. To establish the LoD, five samples containing low levels of analyte (positive clinical samples diluted by known negative clinical samples where the mean values are in the range of LoD or known negative samples spiked with the low positive calibrator) should be tested in duplicate for each lot and day.
This study design will yield 30 replicates of low-level samples per lot. The LoD of the assay is the maximum value of the individually determined LoDs for each lot.
Limit of Quantitation (LoQ):
LoQ is the lowest amount of measurand that can be quantified with stated accuracy. LoQ should be determined according to the recommendations in CLSI EP17 using at least two reagent lots (including two calibrator lots) and performing measurements on at least three days. Four samples with a known low-level concentration of analyte, targeted at LoQ (e.g., known negative samples spiked with the low positive calibrator) should be tested in triplicate for each lot and day.
The LoQ of the assay is the maximum value of the individually determined LoQs for each lot. We recommend the accuracy goal to be set as within-laboratory precision of 20% coefficient of variation (CV). Bias at LoQ based on the assigned values of low calibrators should be set at ≤15%.
Linearity (using clinical samples):
The purpose of the linearity study is to demonstrate the ability of a test (within a given interval) to provide results that are directly proportional to the concentration (amount) of the measurand present in the test sample. Evaluation of assay linearity should be performed according to the recommendations in CLSI EP06, “Evaluation of the Linearity of Quantitative Measurement Procedures: A Statistical Approach; Approved Guideline.” Because performance characteristics can change when sample types have substantially different matrix characteristics, the samples used to determine the linear range of the analytical output should be similar to the samples that will be used during clinical testing. We recommend using at least two clinical samples to generate two linearity sample sets that span the entire claimed measuring interval. Dilutions of each clinical sample should be made using a negative matrix (e.g., if serum and plasma are the sample matrices used, evaluating only serum or plasma may be appropriate) to generate a sample set that spans a range 10 - 20% wider than the expected linear range. At a minimum, five dilutions should be within the linear range. Each diluted sample should be measured using the candidate test to generate at least 4 measurements. If it is not feasible to obtain clinical samples with concentrations of natural SARS-CoV-2 antibodies above the upper limit of the claimed measuring interval, in addition to using at least two clinical samples to generate two separate linearity sample sets, a high-concentration calibrator should be used to generate a high sample that is above the upper limit of the claimed measuring interval. This high sample that is above the upper limit of the claimed measuring interval should then be used to prepare a third linearity sample set in the same manner as the first two linearity sample sets previously described. If the test is intended to be used with venous whole blood samples (i.e., in addition to use with plasma and/or serum), linearity of the assay with venous whole blood samples should also be evaluated separately. Deviation from linearity for each dilution is the difference between the mean values of replicates and the best fitted straight line. All deviations from linearity should be ≤15%.
Precision:
Evaluation of precision of the assay should be performed according to CLSI EP05, “Evaluation of Precision of Quantitative Measurements Procedures; Approved Guideline- Third Edition.” For assays that are not being performed in a single laboratory, a two-site precision study should be conducted. For the precision study design, we recommend the following:
Samples assessed should include a sample close to the assay cutoff, in the expected clinical range, and close to the upper limit of the measuring interval. It is preferable to use clinical samples for this evaluation, although dilutions/contrived samples may be appropriate for measurements at the lower limit. Negative and positive controls used with the assay should also be assessed in the precision study.
Use at least one instrument at each site and a single lot of calibrator/reagent lot. We recommend using different calibrator/reagent lots at each site.
Testing should be performed on three days, two runs per day, and two replicate measurements per run.
Repeatability, between-run, between-day, within-laboratory, between-site precision, and reproducibility should be reported.
Within-laboratory precision is expected to be ≤20%.
Interference:
[Please describe the protocol, and provide results, from an appropriately designed interference study to evaluate the performance of the assay in the presence of high concentrations of potential interfering substances (e.g., conjugated bilirubin, unconjugated bilirubin, lipids/triglycerides, hemoglobin, and protein (albumin).]
To evaluate the effect of potentially interfering substances on the performance of the test, for each potential interferent listed above, we recommend testing a minimum of three individual samples: one sample negative for SARS-CoV-2 antibodies and two samples positive for SARS-CoV-2 antibodies. The positive samples should contain the SARS-CoV-2 antibodies the device is intended to detect at two different levels: one sample should contain SARS-CoV-2 antibodies at slightly above the lower limit of the measuring interval and the other should contain SARS-CoV-2 antibodies at slightly below the upper limit of the measuring interval, (for details, see CLSI EP07, “Interferent Testing in Clinical Chemistry”). As a study control, the same samples should be tested in the absence of the potential interferent.
We recommend you present the results in the following suggested table and calculate the percent difference between the control sample (with no interferent) and the sample with high concentration of the potential interferent.
Potential Interferent (concentration) |
Sample (Analyte Level) |
Mean Numerical Results (Units) |
Result Interpretation |
Percent Difference to Control |
Control (No Interferent) |
Negative |
|
|
|
Control (No Interferent)
|
Lower limit of AMI* |
|
|
|
Control (No Interferent)
|
Upper limit of AMI* |
|
|
|
Hemoglobin (10 mg/mL) |
Negative |
|
|
|
Hemoglobin (10 mg/mL) |
Lower limit of AMI* |
|
|
|
Hemoglobin (10 mg/mL) |
Upper limit of AMI* |
|
|
|
* Assay Measuring Interval (AMI)
Biotin Interference: Biotin may interfere with laboratory tests that incorporate the free biotin-streptavidin capture method in their test methodology.20,21 If the test includes use of biotin, we recommend testing up to 3500 ng/mL with negative and positive samples at the same analyte levels described for potentially interfering substances above. If biotin interference is observed, please provide information to show that you have adequately mitigated this risk.
CRM Accuracy Study with Recovery and Linearity Analysis
This study should be performed using spiking material that is the closest to the CRM in the traceability hierarchy (for example, primary/master calibrators) taking into consideration volume requirements and availability. This is referred to as
“Closest to CRM” (CCRM) in the following recommendations.
It is generally not appropriate to use an authorized test traceable to the CRM instead of using the CCRM to assign values to the candidate test’s calibrators or to support traceability.
To evaluate analytical accuracy for quantitative assays, we recommend conducting a single study to demonstrate the recovery and linearity of the candidate test using the CCRM.
Study Design:
Dilutions of the CCRM should be made using at least two samples collected before the COVID-19 pandemic (collected prior to December 2019) .
Because performance characteristics of the candidate test may be different between sample types that have substantially different matrix characteristics, the negative samples used to make dilutions of the CCRM should be similar to the samples that will be used during clinical testing.
Each negative sample should be a pool of samples from individuals confirmed SARS-CoV-2 antibody negative by the candidate test and the two pools should include samples from different individuals (e.g., Pool 1 should include samples from individuals 1, 2, and 3 while Pool 2 should include samples from individuals 4, 5, and 6). These pools of negative samples should be used to prepare intermediate level samples with the CCRM (either by spiking multiple aliquots of the negative sample with a known amount of CCRM or creating serial dilution). We recommend testing the negative samples using the candidate test prior to preparing the intermediate level samples of CCRM to confirm the samples are negative for SARS-CoV-2 antibodies.
Samples generated from the CCRM dilutions should span the range of the analytical measuring interval for the candidate test. The range of the numeric values of the diluted CCRM samples should be 5%-20% wider than the entire expected analytical measuring interval, taking into consideration the precision of the candidate test. The most diluted level should be below the LoQ.
At a minimum, five intermediate level samples should be within the linear range of the candidate test.
Each intermediate level sample should be measured using the candidate test to generate at least 6 measurements (replicates). If calibrators are part of the candidate test kit, using one reagent/calibrator lot may be appropriate. If calibrators are provided separately, you should evaluate two reagent kit lots and two calibrator lots for a total of four reagent/calibrator lot combinations.
If the test is intended to be used with serum and plasma, and matrix equivalency between the two sample types has been demonstrated, FDA generally considers evaluating only serum or plasma to be appropriate . If the candidate test is intended to be used with venous whole blood samples (i.e., in addition to use with plasma and/or serum), recovery and linearity of the assay with venous whole blood samples should also be evaluated separately.
Data Analysis to evaluate Recovery of the CRM:
We recommend you present the results of the recovery studies in units based on the CRM, as suggested in the Table below.
For each level, you should calculate:
• the average result for the 6 replicates;
• standard deviation (SD) and %CV as SD/average
• recovery =(average)/target x 100
Table: Recovery Data Analysis
Level |
Target concentration, in the CRM Units (e.g., BAU/mL) |
Rep1 |
Rep2 |
Rep3 |
Rep4 |
Rep5 |
Rep6 |
Average |
SD |
%CV |
Recovery |
1 |
|
|
|
|
|
|
|
|
|
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
3 |
|
|
|
|
|
|
|
|
|
|
|
4 |
|
|
|
|
|
|
|
|
|
|
|
5 |
|
|
|
|
|
|
|
|
|
|
|
6 |
|
|
|
|
|
|
|
|
|
|
|
7 |
|
|
|
|
|
|
|
|
|
|
|
Recovery should be between 90%-110%.
Data Analysis to evaluate Linearity with the CRM:
The
purpose of the CRM linearity study is to demonstrate the ability of a
test (within a given interval) to provide results that are directly
proportional to the concentration (amount) in the tested sample using
the CCRM.
Deviations from linearity for each level should be calculated as the
difference between the average of replicates and the best fitted
straight line. For more details, see recommendations in CLSI EP06,
2nd ed. “Evaluation of Linearity of Quantitative Measurement
Procedures” (2020).
All deviations from linearity should be ≤10%.
Cutoff of the Candidate Test in the CRM Units Study:
This study should use CCRM (see section J.6.g above).
For quantitative candidate tests reporting results in CRM Units
(e.g., BAU/mL), you should provide the cutoff for positive and
negative results (assay cutoff) and C9522
established in the CRM Units (e.g., BAU/mL) taking into consideration
the imprecision of the candidate test near the cutoff.
Your
test may fall within either of the following scenarios:
Scenario 1: If you have already established the cutoff of your device in device units (hereafter “Device Units”) (e.g., the device is already authorized with a semi-quantitative claim with internal or arbitrary units such as Index Values, AU/mL, etc.), the purpose of the study is to determine the relationship between the candidate test’s Device Units and the expected numerical values in the CRM Units (e.g., BAU/mL). This relationship should be used to describe the device cutoff in the CRM Units (e.g., BAU/mL) and to establish C95 in CRM Units (e.g., BAU/mL).
Scenario 2: If you are in the process of establishing the cutoff of the candidate test using appropriate clinical samples, the cutoff should be established directly in CRM Units (e.g., BAU/mL) and the purpose of this study is only to establish C95 in the CRM Units (e.g., BAU/mL).
Study design (for either scenario)
Serial dilution should be used to generate at least 5 levels of the CCRM, including dilutions above and below the cutoff of the candidate test. Out of the 5 dilution levels, at least 3 dilutions should be around the cutoff (either in Device Units as in scenario 1 or in the CRM units as in scenario 2) where the expected percent of positive results is larger than zero and less than 100% (including at least 1 dilution with zero percent positive and at least 1 dilution with 100% positive).
SARS-COV-2 negative clinical samples used in the dilutions should be samples collected before the COVID-19 pandemic. We recommend testing the negative samples using the candidate test prior to preparing the serial dilutions of the CCRM to confirm the samples are negative for SARS-CoV-2 antibodies.
CCRM dilution preparations should cover concentrations ±50% from the cutoff. For example, if the cutoff is established at 50 Device Units, then CCRM dilutions from 25 Device Units to 75 Device Units should be evaluated.
The dilutions should be prepared by spiking the CCRM into serum. If there are multiple plasma intended use sample types (e.g., lithium heparin, EDTA, etc.) and matrix equivalency has been demonstrated between the different plasma sample types and serum, the most frequently used serum or plasma type should be evaluated. If matrix equivalency between the plasma sample types and serum has not been demonstrated, this study should be performed in each plasma sample type as well as in serum.
Each sample (i.e., each prepared dilution) should be measured with the candidate test using at least 6 replicates.
Each dilution preparation should be tested with the candidate device separately with at least two kit lots (two reagent and two calibrator lots) .
Data Analysis
The following characteristics of the candidate test with regard to the cutoff and C95 in the CRM units should be provided:
The range of the cut-offs in the CRM Units (e.g., BAU/mL) observed across all reagent lots in the study (for each matrix, if applicable);
The C95 value in the CRM units (e.g., BAU/mL) of the candidate test corresponding to the highest C95 observed per lot (for each matrix, if applicable).
Data Analysis - Scenario 1: (the device cutoff has already been established in Device Units)
For each lot, we recommend that you provide data as suggested in the Table below:
Table: Cutoff of the Candidate Test CRM Units Study - Summary of the Data
Level |
Target concentration in the CRM Units (e.g., BAU/mL) |
Replicate |
Numeric value in the Device Units (e.g., AU/mL) |
Qualitative Results (Positive or Negative) |
1 |
|
1 |
|
|
2 |
|
|
||
3 |
|
|
||
4 |
|
|
||
5 |
|
|
||
6 |
|
|
||
2 |
|
1 |
|
|
2 |
|
|
||
3 |
|
|
||
4 |
|
|
||
5 |
|
|
||
6 |
|
|
For each lot, you should calculate the average of the replicates for each level. We recommend you present results as suggested in the Table below and plot the data as described below.
Table: Cutoff of the Candidate Test in the CRM Units (e.g., BAU/mL) Study - Percent of Positive Results
Level |
Target concentration in the CRM Units (e.g., BAU/mL) |
Average of 6 replicates in Device Units (e.g., AU/mL) |
Percent of positive results |
1 |
|
|
|
2 |
|
|
|
3 |
|
|
|
4 |
|
|
|
5 |
|
|
|
6 |
|
|
|
7 |
|
|
|
The plots showing the relationship between the average of numeric results from the candidate test, expressed in Device Units, and the target numerical values in the CRM Units (e.g., BAU/mL) should be provided as indicated below:
You should plot the CCRM target concentrations in the CRM Units (e.g., BAU/mL) (X-axis) vs average value of all replicates in Device Units (Y -axis). You should perform a visual analysis of whether a linear model is appropriate. If a linear model is appropriate, an ordinary or a weighted least squares regression analysis should be performed. Using the linear regression equation Y=A*X+B, you should calculate the cutoff of the candidate test in the CRM units (e.g., BAU/mL), per lot, as:
Cutoff of the candidate test in the CRM Units (e.g., BAU/mL) = (Cutoff in Device Units - B) / A.
The cutoff in Device Units (e.g., 1.0 Index) should be described in the CRM Units (e.g., BAU/mL) for each lot.
You should plot the CCRM target concentrations in the CRM Units (e.g., BAU/mL) (X-axis) vs percent of positive results (Y-axis). You should use Probit23 analysis to estimate C95 of the candidate test for each lot.
Data Analysis - Scenario 2: (the candidate test cutoff is being established in the CRM Units)
For each lot you should provide data as suggested in the Table below:
Table: Cutoff of the Candidate Test in the CRM Units Study - Summary of the Data
Level |
Target concentration in the CRM Units (e.g., BAU/mL) |
Replicate |
Qualitative Results (Positive or Negative) |
1 |
|
1 |
|
2 |
|
||
3 |
|
||
4 |
|
||
5 |
|
||
6 |
|
||
2 |
|
1 |
|
2 |
|
||
3 |
|
||
4 |
|
||
5 |
|
||
6 |
|
For each lot, you should calculate the percent of positive results. We recommend that you present the results as suggested in the Table below and plot the data as described below.
Table: Cutoff of the Candidate Test in the CRM Units Study - Percent of Positive Results
Level |
Target concentration in the CRM Units (e.g., BAU/mL) |
Percent of positive results |
1 |
|
|
2 |
|
|
3 |
|
|
4 |
|
|
5 |
|
|
You should plot the CCRM target concentrations in the CRM units (e.g., BAU/mL) (X-axis) vs percent of positive results (Y-axis) to demonstrate the relationship between the percent of positive results and the target numerical values in the CRM units (e.g., BAU/mL).
You should use Probit24 analysis to estimate C95 of the candidate test for each lot.
UNMET NEED ADDRESSED BY THE PRODUCT
This section will be completed by FDA.
APPROVED/CLEARED ALTERNATIVE PRODUCTS
There is no adequate, approved, and available alternative to the emergency use of the product.
BENEFITS AND RISKS:
This section will be completed by FDA.
FACT SHEET FOR HEALTHCARE PROVIDERS AND RECIPIENTS:
During review, FDA will make available Fact Sheet templates. See examples for authorized tests on our website.25
INSTRUCTIONS FOR USE/ PROPOSED LABELING/PACKAGE INSERT:
[You should include Instructions for Use, Box Labels, Vial Labels, and any other proposed labeling.]
RECORD KEEPING AND REPORTING INFORMATION TO FDA:
As allowed by Section 564(e) of the FD&C Act, FDA may require certain conditions as part of an EUA. FDA generally includes the following record keeping and reporting information requirements in the EUA.
[Test Developer Name] will track adverse events and report to FDA under 21 CFR Part 803. A website is available to report on adverse events, and this website is referenced in the Fact Sheet for Health Care providers as well as through the [Test Developer Name] Product Support website: [Include link to Test Developer’s website.] Each report of an adverse event will be processed according to [Test Developer Name]’s Non-Conformance Reporting Requirements, and Medical Device Reports will be filed with the FDA as required. Through a process of inventory control, [Test Developer Name] will also maintain records of device usage/purchase. [Test Developer Name] will collect information on the performance of the test, and report to FDA any suspected occurrence of false positive or false negative results of which [Test Developer Name] becomes aware. [Test Developer Name] will maintain records associated with this EUA and ensure these records are maintained until notified by FDA. Such records will be made available to FDA for inspection upon request.
This section applies only to
the requirements of the Paperwork Reduction Act of 1995 The
burden time for this collection of information is estimated to
average 34 to 45 hours per response, including the time to review
instructions, search existing data sources, gather and maintain the
data needed and complete and review the collection of information.
Send comments regarding this burden estimate or any other aspect of
this information collection, including suggestions for reducing this
burden to the address to: Department
of Health and Human Services
An
agency may not conduct or sponsor, and a person is not required to Food
and Drug Administration
respond
to, a collection of information unless it displays a currently Office
of Operations
valid
OMB control number. Paperwork
Reduction Act (PRA) Staff DO
NOT SEND YOUR COMPLETED FORM TO THE PRA STAFF EMAIL ADDRESS
1 This template is part of the “Policy for Coronavirus Disease-2019 Tests During the Public Health Emergency (Revised),” available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/policy-coronavirus-disease-2019-tests-during-public-health-emergency-revised.
2Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/policy-coronavirus-disease-2019-tests-during-public-health-emergency-revised.
3 All EUA templates can be found at https://www.fda.gov/medical-devices/coronavirus-disease-2019-covid-19-emergency-use-authorizations-medical-devices/vitro-diagnostics-euas#covid19ivdTemplates.
4 Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/emergency-use-authorization-medical-products-and-related-authorities.
5 Available at https://www.fda.gov/media/146746/download.
6 Available at https://www.cdc.gov/csels/dls/sars-cov-2-livd-codes.html (last accessed on July 7, 2021). Note this website is not controlled by FDA.
7 Available at https://www.hhs.gov/coronavirus/testing/covid-19-diagnostic-data-reporting/index.html (last accessed on July 24, 2021). Note this website is not controlled by FDA.
8 Available at https://www.hhs.gov/sites/default/files/hhs-guidance-implementation.pdf (last accessed on July 24, 2021). Note this website is not controlled by FDA.
9 Available at https://www.hhs.gov/sites/default/files/non-lab-based-covid19-test-reporting.pdf (last accessed on July 24, 2021). Note this website is not controlled by FDA.
10 Available at https://www.fda.gov/medical-devices/postmarket-requirements-devices/quality-system-qs-regulationmedical-device-good-manufacturing-practices.
11 Further information regarding cybersecurity is available at https://www.fda.gov/medical-devices/digital-health/cybersecurity.
12 Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/guidance-content-premarket-submissions-software-contained-medical-devices.
13 Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/general-principles-software-validation.
14 Available at https://www.fda.gov/medical-devices/digital-health-center-excellence/device-software-functions-including-mobile-medical-applications.
15 Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/shelf-software-use-medical-devices#:~:text=Off%2Dthe%2Dshelf%20(OTS,to%20run%20device%2Dspecific%20functions.
16 GISAID is a global science initiative and primary source that provides open-access to genomic data of influenza viruses and the novel coronavirus responsible for COVID-19 (https://www.gisaid.org/).
17 For more information regarding evaluating the impact of viral mutations and variants, please see the FDA guidance “Policy for Evaluating Impact of Viral Mutations on COVID-19 Tests” available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/policy-evaluating-impact-viral-mutations-covid-19-tests.
18 Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/policy-evaluating-impact-viral-mutations-covid-19-tests.
19 Available at https://www.fda.gov/medical-devices/coronavirus-covid-19-and-medical-devices/sars-cov-2-reference-panel-comparative-data.
20 The FDA issued a Safety Communication regarding Biotin interference on November 28, 2017. It is available at https://www.fda.gov/medical-devices/safety-communications/fda-warns-biotin-may-interfere-lab-tests-fda-safety-communication.
21 For more information regarding Biotin interference testing, please see the FDA guidance “Testing for Biotin Interference in In Vitro Diagnostic Devices,” available at
22 C95 is such a concentration that, under different testing conditions, this sample has positive results 95% of the time. Imprecision of a test for samples near the cutoff is described by an interval [C5; C95]. For more details about C95 and C50, see CLSI document EP12-A2 “User Protocol for Evaluation of Qualitative Test Performance; Approved Guideline- Second Edition.”
23 See CLSI EP17, “Evaluation of Detection Capability for Clinical Laboratory Measurement Procedures; Approved Guideline- Second Edition.”
24 See CLSI EP17, “Evaluation of Detection Capability for Clinical Laboratory Measurement Procedures; Approved Guideline- Second Edition.”
25 A list of EUA-authorized tests and their accompanying fact sheets are available at https://www.fda.gov/medical-devices/coronavirus-disease-2019-covid-19-emergency-use-authorizations-medical-devices/vitro-diagnostics-euas.
| File Type | application/vnd.openxmlformats-officedocument.wordprocessingml.document |
| Author | Gerald, Noel |
| File Modified | 0000-00-00 |
| File Created | 2025-12-05 |
© 2026 OMB.report | Privacy Policy