CDRH COVID-19 Diagnostic Templates (Molecular and Antigen) - Molecular Diagnostic Template
0910-0595_CDRH_molecular_template2.docx
Authorization of Medical Products for Use Emergencies
CDRH COVID-19 Diagnostic Templates (Molecular and Antigen) - Molecular Diagnostic Template
OMB: 0910-0595
OMB: 0910-0595
Exp. date 9/30/2025
Contains Nonbinding Recommendations
Template for Developers of Molecular Diagnostic Tests 1
This template provides the Food and Drug Administration’s (FDA) current recommendations concerning what data and information should be submitted to FDA in support of a pre-Emergency Use Authorization (EUA)/EUA request for a SARS-CoV-2 molecular diagnostic test. FDA generally recommends that the following validation studies be conducted for SARS-CoV-2 molecular diagnostic tests: limit of detection (LOD), inclusivity, cross-reactivity, sample stability, and clinical evaluation.
As described in the FDA guidance document Policy for Coronavirus Disease-2019 Tests During the Public Health Emergency (Revised), 2 FDA is providing recommendations in this and other EUA templates regarding testing that should be performed to ensure appropriate analytical and clinical validity, including descriptions of appropriate comparators, for different types of tests. The EUA templates3 are intended to help test developers provide recommended validation data and other information to FDA, but alternative approaches can be used. This template reflects FDA’s current thinking on the topic, and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word should, means that something is suggested or recommended, but not required. For more information about EUAs in general, please see the FDA guidance document: Emergency Use Authorization of Medical Products and Related Authorities.4
Test developers interested in pursuing an EUA may submit a pre-EUA to begin discussions with the FDA or may submit an EUA request to Covid19DX@fda.hhs.gov.
FDA recommends that all developers of molecular SARS-CoV-2 tests include the Molecular EUA Template Cover Sheet5 when submitting their EUA request to Covid19DX@fda.hhs.gov to help streamline the routing, triage, and review of EUA requests.
GENERAL INFORMATION ABOUT THIS TEMPLATE
Text highlighted in yellow [Text] should be completed by the test developer as applicable to their specific test. Text in bold outlines the FDA’s additional recommendations for the developers’ consideration when completing the suggested information in each section.
Not all portions of this template may be relevant for all developers/tests. FDA recommends developers complete all portions that are relevant to facilitate a streamlined review.
This template addresses tests intended for use with respiratory samples and saliva; if you are considering other sample types, please contact FDA at CDRH-EUA-Templates (Covid19DX@fda.hhs.gov) to discuss your validation strategy.
A test authorized under an EUA is only authorized for emergency use while the EUA is in effect.
We may update the template as appropriate as we learn more about COVID-19 and gain experience with the EUA process for these kinds of tests.
A developer that has provided data to the FDA may grant a right of reference to other developers, either broadly or to individual developers, to leverage that data. A right of reference provides a developer the ability to rely upon, and otherwise use, existing information in one regulatory submission for the purpose of supporting a different regulatory submission. In these cases, if the data is applicable to the new developer's test, the new developer may not have to repeat that validation for its submission to the FDA or FDA may recommend only a bridging study. Any developer seeking to leverage data regarding another developer’s EUA-authorized assay must obtain a right of reference from that developer. 6
EXAMPLE TEMPLATE:
Emergency Use Authorization (EUA) request for distribution and/or use of the [test name] for the in vitro qualitative detection of RNA from the SARS-CoV-2 in [add all claimed sample types, e.g., nasopharyngeal/ oropharyngeal swabs, sputa, bronchoalveolar lavage (BAL), etc.] [select appropriate testing population, e.g., from patients suspected of COVID-19 by a healthcare provider or for screening of individuals without symptoms or other reasons to suspect COVID-19.]. Test results should be reported in accordance with local, state, and federal regulations.
[If you plan to include a sample pooling protocol in your instructions for use, please include a brief description of the pooling strategy.]
[If you plan to request authorization for screening with serial testing, please refer to the Supplemental Template for Developers of Molecular and Antigen Diagnostic COVID-19 Tests for Screening with Serial Testing and include any relevant information.]
[If you plan to request authorization to test samples collected with a home specimen collection kit, please refer to the Home Specimen Collection Molecular Diagnostic Template and include any relevant information.]
Specific nucleic acid sequences from the genome of the SARS-CoV-2 [please specify the targeted gene(s) of the pathogen].
[Official name, address, and contact information (including phone number and email address) of applicant and primary correspondent.]
D. PROPRIETARY AND ESTABLISHED NAMES
Proprietary Name - [test name]
Established Name - [test name]
The [test name] test is not cleared, CLIA waived, approved, or subject to an approved investigational device exemption.
[If the test has been previously reviewed in an EUA request or pre-EUA submission, please provide the submission number.]
Panel Code: MI for Microbiology tests
Review Group: Division of Microbiology Devices/VIR1
Product Code:
QJR (for SARS-CoV-2 only tests)
OR
QLP (for multi-analyte tests that include SARS-CoV-2)
The proposed IU will be finalized based on, among other things, the data provided and recommendations from Public Health authorities at the time of authorization – example text is provided below for a qualitative molecular test that detects organism RNA but may be adapted according to the specific emergency situation addressed by the device, proposed intended use population, testing sites, or performance characteristics.
The [test name] is a [specify test technology such as, real-time RT-PCR test] intended for the [presumptive] qualitative detection of RNA from SARS-CoV-2 in [describe all the sample types that were evaluated, e.g., nasopharyngeal, nasal, and oropharyngeal swab samples and lower respiratory tract, BAL, sputum] [If your test is intended for testing multiple respiratory pathogens, please list the specific analytes detected by your test.] [describe intended use population, e.g., from individuals suspected of COVID-19 by their healthcare provider or for screening of individuals without symptoms or other reasons to suspect COVID19 infection.]. Testing is limited to [laboratories certified under the Clinical Laboratory Improvement Amendments of 1988 (CLIA), 42 U.S.C. §263a, that meet the requirements to [insert testing complexity, e.g., moderate complexity, high complexity, or waived tests. This test is authorized for use at the Point of Care (POC), i.e., in patient care settings operating under a CLIA Certificate of Waiver, Certificate of Compliance, or Certificate of Accreditation.].
[Describe the sample pooling approach, as appropriate]. This test is also for the qualitative detection of nucleic acid from SARS-CoV-2 in pooled samples containing up to [maximum number] individual [type of sample and collection method], where each specimen is collected using [type of sample and collection method]. Negative results from pooled testing should not be treated as definitive. If a patient’s clinical signs and symptoms are inconsistent with a negative result or if results are necessary for patient management, then the patient should be considered for individual testing. Specimens included in pools with a positive or invalid result must be tested individually prior to reporting a result. Specimens with low viral loads may not be detected in sample pools due to the decreased sensitivity of pooled testing. For specific patients whose specimen(s) were the subject of pooling, a notice that pooling was used during testing must be included when reporting the result to the clinician or healthcare provider.
Results are for the identification of SARS-CoV-2 RNA. The SARS-CoV-2 RNA is generally detectable in [name sample type, e.g., upper respiratory] during the acute phase of infection. Positive results are indicative of the presence of SARS-CoV-2 RNA; clinical correlation with patient history and other diagnostic information is necessary to determine patient infection status. Positive results do not rule out bacterial infection or co-infection with other viruses. The agent detected may not be the definite cause of disease.
Laboratories within the United States and its territories are required to report all test results to the appropriate public health authorities.
The [test name] is intended for use by [include intended user, e.g., qualified, and trained clinical laboratory personnel specifically instructed and trained in the techniques of RT-PCR and in vitro diagnostic procedures].
The [test name] is only for use under the Food and Drug Administration’s Emergency Use Authorization.
Depending on the performance and the populations studied in the clinical evaluation, additional limitations may be recommended.
For prescription use only
For in vitro diagnostic use
For Emergency Use Authorization only
The [test name] test is to be used with the [list all RT-PCR instruments, software, and automated extraction instruments, other applicable instrumentation, etc.].
If your test system includes an instrument, the instrumentation manual should be submitted as part of the EUA request. If your test system includes an instrument that was not previously cleared, approved, or authorized by FDA, please see additional discussion in the Product Manufacturing section and note that additional labeling information may be discussed during the EUA review.
G. DEVICE DESCRIPTION AND TEST PRINCIPLE
Please provide a device description. The example provided below applies to fluorescence based real-time reverse-transcriptase-polymerase chain reaction (RT-PCR) tests for detection of organism RNA. Please modify the example text as appropriate for tests that use a different test principle. For new technologies, FDA may request additional information so we can adequately assess the known and potential risks and benefits associated with the device.
[Describe the technology of the test and how this technology works to identify the measurand (i.e., the test principle), the instruments/reader employed/required to perform the test from sample collection to result (include all claimed extraction and PCR detection instruments), and the sample types for which the performance of the test has been established. If applicable, list all primer and probe sets, briefly describe what they detect, and include the nucleic acid sequences. Please indicate if the test uses biotin-Streptavidin/avidin chemistry in any of the steps for coupling reagents.]
The [test name] is a real-time reverse transcription polymerase chain reaction (RT-PCR) test. The SARS-CoV-2 primer and probe set(s) is designed to detect RNA from the SARS-CoV-2 in [list all the sample types] from patients suspected of COVID-19 by their healthcare provider.
[List and describe in detail all the steps of the test sequentially from sample collection to assay report.]
[Step one]
[Step two]
Etc.…]
Nucleic acids are isolated and purified from [samples] using [please describe the method(s) of extraction (please specify the sample input volume for extraction and/or test, the nucleic acid elution volume and whether isolation/purification is manual and/or automated)]. The purified nucleic acid is reverse transcribed using [enzyme mix/kits – please specify the input volume of purified nucleic acid added to the RT-PCR reaction mix] into cDNA which is then subsequently amplified in [please describe the instrument(s) and enzyme mix]. In the process, the probe anneals to a specific target sequence located between the forward and reverse primers. During the extension phase of the PCR cycle, the 5’ nuclease activity of Taq polymerase degrades the probe, causing the reporter dye to separate from the quencher dye, generating a fluorescent signal. With each cycle, additional reporter dye molecules are cleaved from their respective probes, increasing the fluorescence intensity. Fluorescence intensity is monitored at each PCR cycle by [please describe the detection instrument(s)].
[List all control materials (provided with the test kit and/or required but not provided with the test kit, e.g., sold as a separate kit) and describe what they are, how they are expected to work, where in the testing process they are used, and the frequency of use. If a control is commercially available, provide supplier’s name and catalog number or other identifier; if your device relies on external controls that are manufactured by a third party please note that these controls should also be validated within your analytical and clinical studies described below in Section J.]
Controls that will be provided with the test kit include:
An external positive template control is needed to [describe need] and is used [describe use – please specify the concentration of the positive control relative to the LoD of your test (note that ideally the positive control concentration should be such that it is close to the LoD of your test) and specify frequency of use.]
An external negative control is needed to [describe need] and is used [describe use – please specify the composition of the negative control and specify frequency of use.]
An extraction control [describe control] is needed to [describe need] and is used [describe use – please also specify frequency of use]. Please note that if the positive control is taken through the entire sample processing procedure, including the extraction, then a separate extraction control is not required.
A [other (e.g., sample adequacy, internal, etc.)] control is needed to [describe need] and is used [describe use – please specify the composition of the control and specify frequency of use]
Controls that are required but not provided with the test kit include [describe control – provide recommended sources of the control materials – either a separate control kit for purchase that you the applicant develops or a control material that can be purchased from a third party]. This/these control(s) is/are needed to [describe need] and is used [describe use – please also specify frequency of use].
Please note that any control used with your device (provided with the kit or not) should be validated in the context of your analytical and clinical studies (i.e., your studies should include use of these controls). In instances where control material is not readily available through 3rd party vendors, FDA recommends that you include suitable control material with your device. External control materials are considered particularly important when good manufacturing practice (GMP) requirements are waived, and reagent stability studies are limited.
Test Result Reporting:
All test results are to be reported to healthcare providers and relevant public health authorities in accordance with local, state, and federal requirements, using appropriate LOINC and SNOMED codes, as defined by the Laboratory In Vitro Diagnostics (LIVD) Test Code Mapping for SARS-CoV-2 Tests7 provided by the Centers for Disease Control and Prevention (CDC). Core diagnostic data elements8 are to be collected for all tests, which have been defined by the Department of Health and Human Services (HHS), along with technical specifications for implementation for lab-based9 and non-lab-based10 tests.
All test controls should be examined prior to interpretation of patient results. If the controls are not valid, the patient results cannot be interpreted. [Appropriate control interpretation criteria should appear in your product labeling. Please describe if a Ct (cycle threshold) cutoff is used as part of your testing algorithm and/or if the end user is required to review curves before final result interpretation. Although not typical for molecular-based tests, if the test result involves the use of an algorithm/calculation, for example a ratio value, when determining the final patient test result, please include a detailed description and any additional calibration materials that may be required.]
[Describe in detail the expected results generated, including acceptance criteria, for all the controls described in Section G above. Describe the measured values (if applicable) for valid and invalid controls and outline the recommended actions the laboratory should take in the event of an invalid control result.]
[Describe
when clinical sample test
results should be assessed and outline the criteria for test
validity.]
Example
text: Assessment
of
[test
name] results
should be performed after the positive and negative controls have
been examined and determined to be valid and acceptable. If the
controls are not valid, the patient results cannot be interpreted.
[Clearly indicate how to interpret numeric test values (if applicable) as positive or negative for presence of SARS-CoV-2. Indicate if the end user is required to review curves before final result interpretation and, if applicable, how to identify indeterminate/inconclusive/equivocal results. When applicable, we recommend providing a table clearly describing the possible combinations of test result values for each primer/probe set. Describe how they should be combined into a final interpretation of the result for your test. If the test produces an equivocal or indeterminate result, please indicate what follow-up testing/process should be conducted, if applicable.]
The [test name] has been validated using only the components referenced in this request and will not be changed after authorization without prior concurrence from the FDA.
Overview of Manufacturing and Distribution:
The product will be manufactured at [test developer’s name and FDA registration number (if applicable)] by [test developer’s name] personnel consistent with practices for the production of [types of devices] based on [type of quality system (e.g., 21 CFR 820 or ISO13485)]. Material manufactured by [test developer’s name] may be bottled and kitted by [packager name] manufacturing facility.
The current manufacturing capabilities include the ability to manufacture approximately [please insert the approximate number of tests/kits that can currently be manufactured per week at the manufacturing facility] products per week for distribution in the United States, however, in the event of a surge in demand this could be increased to [please insert the approximate maximum number of tests/kits that could potentially be manufactured per week at the manufacturing facility if there was a surge in demand] product per week within a [please specify in weeks/months the expected timeframe required to increase product production if conditions warrant] timeframe.
Under an EUA, certain sections of the 21 CFR Part 820 Quality System Regulation (QSR) requirements may be waived for an authorized produced during the duration of the EUA, but FDA recommends that test developers follow comparable practices as much as possible, even if such requirements are waived. Please see recent letters of authorization for examples of which QSR requirements have been required.
[Please specify any instruments or other components of your test which are labeled as research use only (RUO) or are otherwise not labeled with the statement “For In Vitro Diagnostic Use” or a symbol found in a standard to the same effect.]
For distributed tests (i.e., tests intended to be performed in more than one laboratory location), that use an RUO instrument, please provide the following information, as applicable:
FOR AN RUO INSTRUMENT WHERE THE EUA REQUESTER IS NOT THE MANUFACTURER OF THE INSTRUMENT:
Please include in the instructions for use found in your test’s labeling, appropriate procedures, including acceptance criteria, that laboratory customers should follow to qualify the performance of the RUO instrument prior to use with your test.
These procedures could include wet testing of quantitated test material with your test, or confirmation that key specifications of the instruments that are applicable to your test are within an acceptable range. The quantitated virus material could either be positive control material included with your kit or commercially available positive virus control material. If commercially available material is not labeled with the statement “For In Vitro Diagnostic Use” or a symbol found in a standard to the same effect, then you should qualify lots of this material in-house and have a mechanism to notify laboratory customers which lots are appropriate to use for qualification (i.e., posting on a website). For the qualification protocol, you should include a recommendation to test multiple dilutions of virus material with your test, with, at minimum, 3 replicates per dilution. There should be at least one dilution near the LoD (i.e., within 3x LoD) of your test. The protocol should outline the acceptance criteria for each dilution tested.
[Please also provide the following labeling documentation with your request:
A "For Emergency Use Authorization only" label that users can affix to the instrument after it has been qualified. This can be provided as an Appendix in the assay instructions for use.
Please ensure that your test’s labeling either reproduces the parts of the instrument operating manual that are relevant to run your test or references the relevant sections of the manual.]
FOR AN RUO INSTRUMENT WHERE THE EUA REQUESTER IS THE MANUFACTURER OF THE INSTRUMENT:
[Please either provide the qualification protocol as described above or the following information to demonstrate your instrument meets the minimum quality system requirements for authorization:
The ISO 13485 certificate for the site where your instrument is manufactured.
A document mapping out the parts of your quality system that fulfill each of the following 21 CFR part 820 requirements:
Subpart H (Acceptance Activities, 21 CFR 820.80 and 21 CFR 820.86),
Subpart I (Nonconforming Product, 21 CFR 820.90), and
Subpart O (Statistical Techniques, 21CFR 820.250).
Please provide the following labeling documentation with your request:
A “For Emergency Use Authorization only” label that the users can affix to the instrument after it has been qualified. This can be provided as an Appendix in the assay instructions for use.
The instrument operating manual. Please note that the manual should not include any unapproved, uncleared, or unauthorized uses.
An instrument manual addendum that will be distributed along with your EUA test kit. The addendum may have the following format:]
Instrument Operation Manual Addendum:
For emergency use authorization only with the [test name].
The [test name] is authorized for use under the US Food and Drug Administration (FDA) Emergency Use Authorization (EUA) with the [name of instruments] for the [presumptive] qualitative detection of RNA from SARS-CoV-2 [intended use of test]. Refer to the [test name] instructions for use for additional information [provide hyperlink].
This instrument operation manual addendum applies to the instruments listed in Table below that are authorized for use with the [test name].
Table: Instruments Authorized for Emergency Use Only with the [test name]
Catalog Number |
Product Name |
|
|
|
|
|
|
|
|
Warnings:
This product has not been FDA cleared or approved; the product has been authorized by FDA as part of [test name] under an EUA for emergency use only by authorized laboratories certified under the Clinical Laboratory Improvement Amendments of 1988 (CLIA), 42 U.S.C § 263a.
This product has been authorized only for the detection of nucleic acid from SARS-CoV-2, not for any other viruses or pathogens.
The emergency use of this product is only authorized for the duration of the declaration that circumstances exist justifying the authorization of emergency use of in vitro diagnostics for detection and/or diagnosis of COVID-19 under Section 564(b)(1) of the Federal Food, Drug, and Cosmetic Act, 21 U.S.C. § 360bbb-3(b)(1), unless the declaration is terminated, or authorization is revoked sooner.
Components manufactured by [test developer’s name and FDA registration number (if applicable)] and supplied with the test include:
[List all components and other materials/information included with your test, including a description of the primers and probes, volumes, concentrations, quantities, buffer components, etc.]
Table: Example: Kit Components & Other Materials/Information
Kit Components & Other Materials/Information |
Main Reagents Composition/ Matrix |
Concentration/ Quantity/Volume |
Manufacturer |
Test Cassette with test strip |
|
|
|
Negative control |
|
|
|
Positive control |
|
|
|
Calibrators |
|
|
|
Sample buffer (bottle) |
|
|
|
Transfer pipette |
|
|
|
Instructions for Use leaflet |
|
|
|
Packing materials |
|
|
|
Others, as applicable |
|
|
|
[List all components and other materials/information (e.g., instruments, reagents) not included with the test that must be supplied by the user to perform the test, with specific supplier names and catalog numbers or other identifiers for obtaining the components. Please include here all specific consumables that were validated for use with your device, that are not interchangeable with other products and that are needed to guarantee device performance as established in the EUA validation studies listed in Section J below.]
If you are introducing a system onto the market that has not been previously reviewed by FDA, we recommend providing evidence that the software has been validated to ensure that:
The inputs and outputs of the software are appropriate to fulfill the system and assay requirements;
All expected inputs produce the expected outputs for all functions critical for system operation; and
The system will be provided to the customer free of defects, or defects will be known and mitigated.
If this evidence is not available prior to authorization and the software and hardware have been designed and developed in a manner consistent with current GMPs (for additional information, please see the discussion of “Quality System Regulation/Medical Device Good Manufacturing Practices,” on the FDA website11), additional software validation documentation may be incorporated into the conditions of authorization. If changes which impact assay performance or safety and effectiveness of the system are needed to address validation failures post-authorization, an EUA supplement may be required under the conditions of authorization.
Below are examples of tables for providing system specific information and your evidence that specifications have been met (e.g., hazard analysis). Text in the tables is provided as an example only. [Please provide thorough functional descriptions of system software and instrumentation specifications needed to support the intended use of the test and provide evidence that specifications have been fulfilled.]
System specifications and validation example
Critical specifications: Description of the specification |
Evidence that the design of the system can fulfill the specification. This column should consist of system-level validation data. |
Optical system of each instrument sent to a user has sufficient dynamic range to appropriately differentiate between positive and negative test results |
|
Software displays appropriate result during test run |
|
If reader stores test result, software accurately stores and retrieves test results |
|
System has a defined lifetime where the user can expect the system to maintain performance as stated in the label |
|
Etc. |
|
Hazard analysis examples
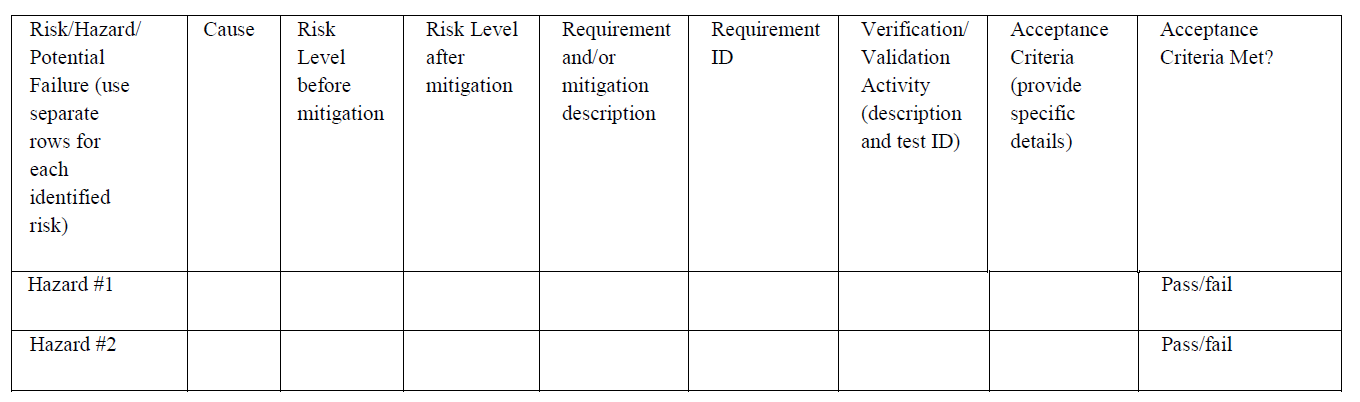
-
ID
Hazard
Adverse Effect
Severity
Potential causes of hazard
Risk mitigation measure
Risk of experiencing the hazard after mitigation
1
Invalid result
Delay in returning test result
Low
User inserts cartridge incorrectly
Labeling noting correct orientation
Low
2
False result
Wrong result returned to user
High
Incorrect alignment of test strip and optics; test strip inserted in the wrong orientation
Mechanical design of reader input slot
Moderate
3
False negative result
Wrong result returned to user
High
User reads test strip too early; incubation time not sufficient
Labeling noting correct incubation time
Moderate
4
False result
Wrong result returned to user
High
Incorrect alignment of test strip and optics; control line misinterpreted
Software interprets data from optical system identifying a valid/invalid control
Moderate
5
False result
Wrong result returned to user
High
Control reaction intensity is misinterpreted
Software interprets data from optical system identifying a valid/invalid control
Moderate
6
False result
Wrong result returned to user
High
Analyte reaction intensity is misinterpreted
Software interprets data from optical system identifying a valid/invalid control
Moderate
If applicable to your test, FDA recommends the following evaluations be performed and documentation kept on file. If not completed by the time of authorization, these evaluations may be required in a condition of authorization.
You should evaluate the cybersecurity of your system to ensure user and patient safety in the intended use environment;12
You should complete validation of all systems and software to ensure that all functions of the system perform as labeled. For more information on system validation please see the following FDA guidance documents and resources:
[If you are introducing a system onto the market which has not been previously reviewed by the FDA, please describe how you addressed basic safety hazards such as electrical hazards (e.g., electrical shock to the operator and/or patient), fire hazards, and mechanical hazards.] We recommend that you consult the general requirements for basic safety, as indicated in International Electrotechnical Commission (IEC) 60601-1 (Medical electrical equipment – Part 1: General requirements for basic safety and essential performance). IEC 60601-1 is a standard that specifies the general requirements for basic safety and essential performance. IEC 60601-1 defines basic safety as freedom from unacceptable risk directly caused by physical hazards when medical electrical equipment is used under normal condition and single fault condition.
Electromagnetic Compatibility (EMC) Testing (if applicable)
We recommend that EMC testing be conducted on any assay that uses a battery or power source. [Please provide FDA with any standards that were followed for EMC testing.] We recommend that you perform EMC testing according to the International Electrotechnical Commission (IEC) 60601-1-2 Edition 4.0:2014. [If you perform EMC testing to a different standard or use alternate methodologies to evaluate EMC, please provide a test plan, test report, acceptance criteria, and risk analysis to support your approach.]
[Briefly describe current sample throughput testing capacity, the total time required to perform the test (from clinical sample collection to result), and the number of tests that can be performed per day (8-hour shift), excluding controls and calibrators, as applicable. Please provide the number of kits you can manufacture per day/week for distribution in the United States.]
The product will be distributed by [please describe the distribution plan for the product and list all current US distributors].
Reagent Stability
[Briefly describe the stability test plan for [test name] reagents and include any accelerated stability information, if available.] Reagent stability studies generally do not need to be completed at the time of EUA issuance; however, the study design should be agreed upon during interactive review and the stability studies started immediately following authorization, if not before. You should consider the following recommendations when designing your stability study:
For EUAs you may follow the current FDA recognized “Clinical Laboratory Standards Institute (CLSI) Standard EP25 – Evaluation of Stability of In Vitro Diagnostic Reagents; Approved Guideline” when evaluating the suitability of stability study designs. If you are planning to pursue a clearance or approval for your device, we recommend discussing with FDA in more detail your stability design to facilitate potential use of the EUA data in your regular premarket submission.
For all the stability evaluations, you should include your external positive and negative controls and at least one sample, which should be prepared by spiking negative clinical matrix at an analyte concentration of 3-5x LoD of inactivated SARS-CoV-2, not recombinant protein. Please note that use of the positive controls alone is not recommended for reagent stability evaluation because controls are usually formulated at a moderate positive level.
If you are using multiple clinical sample types in which similar LoDs are determined, you should use the most challenging clinical matrix for this study.
You should evaluate at least 5 replicates and, if available, 3 different lots of reagents.
You should design your study to provide data for a timeframe that is about 10% longer than the one to be authorized. For example, 18 months should be supported by stability data out to 20 months and 7 days should include stability data out to 8 days.
FDA considers 15-30°C to represent room temperature conditions. Ideally you should evaluate stability at both 15°C and 30°C; however, for the purposes of the EUA evaluation, 30°C is generally appropriate as the worst-case scenario.
Unopened Kit Shelf-Life Stability:
You should evaluate real-time kit stability studies with unopened kits stored at the claimed storage temperature for your test.
Accelerated stability evaluations for unopened kits can be included for EUA requests to support shelf-life claims up to six months while the real-time studies are on-going. However, please note real-time stability data is generally needed to support regular pre-market submissions.
Unopened Kit Shipping Stability: You should evaluate the anticipated handling and shipping times and temperatures expected for unopened kits under different temperature conditions (e.g., summer, winter). The recommended summer profile is storage at 40℃ for 8 hours and then 22℃ for 4 hours and the recommended winter profile is -10℃ for 8 hours and then 18℃ for 4 hours.
In-use/Opened Kit Stability: Depending on your device, your stability study design should also support in-use stability of the kit reagents once the kit has been opened, e.g., storage at 2-8°C for 7 days. This includes on board stability once reagents have been placed on the instrument (if applicable).
Inverted stability (if applicable): Study should support stability for kits if stored inverted or in the wrong orientation.
Freeze-thaw Stability: If you recommend aliquoting the reagents to meet the end-users needs following the initial thaw this recommendation should be supported by a freeze-thaw stability study, including the specific number of allowed freeze-thaw cycles.
FDA recommendations for analysis of real time stability studies are as follows:
Baseline of the study (t=0 of stability study) should not exceed one month from production;
Clear baselines should be described (e.g., one month from production) for each stability claim under each study;
Claims should be determined based on regression analysis. Any %change (%shift) from time zero (baseline) should be calculated between the target claim and the zero-time as (Ttest-Tbaseline)/ Tbaseline*100 with 95% confidence interval (CI) using the regression equation obtained from plotting the mean values. When formulating your acceptance criteria for evaluating the shift from baseline you should consider the reproducibility of your device. Generally, the shift at the target claim due to storage cannot exceed 10-15%. The target stability is the next to last tested point that was within +/- 10% of time zero; and
Acceptance criteria may differ depending on the reproducibility of your device, the distribution of analyte concentration expected in samples from the intended use population, and the risk of false results to public health.
J. PERFORMANCE EVALUATION
The following validation studies should be performed to support your EUA request. Please note that, particularly for new technologies, FDA may request additional studies so we can adequately assess the known and potential risks and benefits associated with the candidate test. [For each validation study, you should provide a study protocol that includes a detailed, step-by-step description of how samples were prepared and how testing was conducted. You should also include complete study line data in an Excel-compatible format for all validation studies. Such line data should include the following information in individual columns:
coded identifiers for all samples and replicates;
the clinical matrix tested;
the SARS-CoV-2 concentration (applicable to studies using contrived samples);
raw signal output (i.e., cycle threshold (Ct) values) and final results for each distinguishable target for both the candidate test and the comparator test (as applicable); and
for both the candidate test and the comparator test, a final result for each sample/replicate based on the result interpretation algorithm of the test.]
You should determine the LoD of the candidate test utilizing the entire test system from sample preparation and extraction to detection. We recommend spiking quantified inactivated virus (e.g., heat treated, chemically modified, or irradiated virus) into real clinical matrix (e.g., nasal or nasopharyngeal (NP) swabs, bronchoalveolar lavage (BAL) fluid, sputum, etc.). As positive natural clinical samples are generally available, a quantified known positive clinical sample as determined by an EUA- authorized test can be used to create dilutions in clinical matrix for LoD determination. Synthetic RNA is not an appropriate test material for the LoD studies. Respiratory samples collected from SARS-CoV-2 negative individuals can be used as clinical matrix. Collection media without clinical matrix or collection kits that were not used to collect a clinical sample are generally not considered real clinical matrix.
FDA recommends that preliminary LoD be determined by testing a 2-3-fold dilution series of 3 replicates per concentration, and then confirmed with 20 replicates of the concentration determined to be the preliminary LoD. For purposes of this document, the preliminary LoD is the lowest concentration that gives positive results 100% of the time and the final LoD is the lowest concentration at which at least 19 of 20 replicates are positive. The preliminary LoD studies should include at least one concentration that does not yield 100% positive results. If multiple clinical matrices are intended for clinical testing, you should include the results from one representative matrix of each indicated clinical matrix to FDA. The most challenging matrix of the claimed matrices should be tested. For example:
If the candidate test is indicated for testing common upper respiratory tract samples (e.g., nasopharyngeal (NP) swabs, oropharyngeal (OP), swabs, nasal swabs, anterior nasal swabs, mid-turbinate nasal swabs, nasal aspirates, and nasal washes etc.), please submit results from NP swabs as FDA considers this to be the most challenging upper respiratory matrix.
If the candidate test is indicated for testing common lower respiratory tract samples (e.g., tracheal aspirates, sputum, etc.), please submit results from sputum as FDA considers this to be the most challenging lower respiratory matrix.
If the candidate test is indicated for testing both, upper and lower respiratory matrixes, submitting results from sputum samples may suffice to support both upper and lower respiratory matrices.
If the candidate test is indicated for testing alternative samples, such as saliva, oral fluid, buccal swab, etc., please submit results from testing each of the claimed uncommon sample types.
If relevant, FDA recommends that you follow the most current version of the CLSI EP17 “Evaluation of Detection Capability for Clinical Laboratory Measurement Procedures.”
[Please provide your complete LoD study protocol that includes a step-by-step description of how samples were prepared and tested with your device, the specific viral material used to assess the LoD (e.g., irradiated virus), and the LoD (with appropriate units) for your assay. Please provide the line data for the LoD study in an Excel-compatible format. If the assay includes use of an analyzer or application to generate test results, please include the analyzer value for each test replicate.]
LoD studies determine the lowest detectable concentration of SARS-CoV-2 at which approximately 95% of all (true positive) replicates test positive. The LoD was determined by limiting dilution studies using characterized [please describe samples used in the study, e.g., viral stocks].
[List/describe the following in this section:
Titers and strains of the SARS-CoV-2 stocks used for the LoD study and how the organism stocks were prepared and how the titers were determined.
The dilution factor and number of serial dilutions of the characterized SARS-CoV-2 that were tested to determine the LoD.
The starting concentration, dilution factor used to reach target concentration, the volume of negative matrix with inactivated SARS-CoV-2 spiked onto each swab in your LoD study, and the type of dilutant used (e.g., Phosphate Buffered Saline (PBS), saline, etc.) to prepare each replicate in your LoD study. Please note that it is generally not appropriate to prepare samples with your assay reagents (e.g., extraction buffer) nor is it generally appropriate to dilute clinical matrix in VTM if the test is not indicated for use with VTM.]
Serial dilutions of the characterized SARS-CoV-2 made in clinical matrix obtained from individuals who tested negative for SARS-CoV-2 were then tested in [number of replicates] replicates. The lowest concentration at which all [number of replicates] replicates were positive was treated as the tentative LoD for each test. The LoD of each test was then confirmed by testing [number of replicates (at least 20 recommended)] with concentrations at the tentative limit of detection. The final LoD of each test was determined to be the lowest concentration resulting in positive detection of [number of positive replicates (at least 19 out of 20 replicates)]. [Include analysis of LoD results, indicating the final LoD for each test.]
Note: The LoD range finding study should include at least one concentration that achieves 95% detectability of replicates and at least one concentration that achieves less than 95% detectability. Replicates should be interpreted per the result interpretation of your test.
Mutations in the SARS-CoV-2 genome have been identified as the virus has spread. A mutation is an individual genetic change in a SARS-CoV-2 virus sequence when compared with a reference sequence such as Wuhan-Hu1 or USA-WA1/2020. A new virus variant of SARS-CoV-2 has one or more mutations that differentiate it from the wild type or predominant virus variants already circulating in the general population. Variants of SARS-CoV-2 are identified by genomic sequences that contain mutation(s) in the RNA genome, which could result in amino acid substitutions, insertions, and/or deletions in viral proteins. Different variants can result in different phenotypes (e.g., a difference in antigenicity, transmissibility, or virulence). Viral mutations and viral variants could result in altered immunogenicity relative to the originally isolated virus, which could impact the performance of in vitro tests.
Test developers should monitor new and emerging viral mutations and variants that could impact molecular test performance on an ongoing basis. This includes assessing the prevalence of viral mutations in sequence databases (e.g., the GISAID17 database), as mutations observed in these databases at a significant frequency may signify that the mutation is present in an increasing proportion of infected individuals in the U.S. FDA currently considers a significant frequency to be greater than 5% (when considering at least 2000 sequences over a recent period of time, such as the past week, month, or quarter). Monitoring should also include identifying if there are multiple credible reports indicating that a given viral variant (which may have one or more mutations) has the potential to increase virulence, increase transmission, or otherwise increase the public health risk. FDA recommends monitoring on at least a monthly basis in light of the rate of occurrence of mutations and variants and the importance of assessing their impact.
For any viral mutations and variants that are identified as prevalent and/or clinically significant as described above, you should assess whether the resulting predicted amino acid change(s) in the viral proteins are critical to your test design. This may be accomplished via in silico analysis of published SARS-CoV-2 sequences compared to the assay’s primers and probes. If the mutations are found to be critical to your test design, such mutations and variants should be evaluated using clinical (or contrived, as available and as appropriate) samples to assess the impact of the mutation or variant on your test’s performance. The aggregate impact of the mutations should not reduce the clinical performance of the test by 5% or more or decrease the clinical performance point estimates for the test below the clinical performance recommendations described in Section J(10). Please see the FDA guidance document “Policy for Evaluating Impact of Viral Mutations on COVID-19 Tests” for additional discussion regarding monitoring the impact of genetic variants on molecular diagnostic tests.18
FDA also has ongoing monitoring efforts and may identify a viral mutation or variant as clinically significant for which testing with clinical (or contrived, as available and as appropriate) samples would be recommended to assess the impact of the mutation or variant on the performance of your test.
We recommend providing a summary of the strategy used to choose targeted amplification regions and the specific primer and probe regions, as applicable. Developers should document the methodology and results of an in silico inclusivity analysis that establishes the extent to which variation in the SARS-CoV-2 genome may impact sensitivity of test performance.
[Please provide your plan for monitoring for new and emerging SARS-CoV-2 viral mutations and variants on an ongoing basis and for assessing the impact of mutations and variants that have been identified as prevalent and/or clinically significant on the performance of your assay over time.]
[For mutations and variants that have been identified as prevalent and/or clinically significant as part of ongoing monitoring at the time of your EUA request, please provide information on the potential impact of the mutation(s) and variants on your test’s performance or explain how the risk associated with the unknown performance of your device in samples from individuals with the variant(s) can be adequately mitigated.]
Cross-reactivity studies are performed to demonstrate that the test does not react with related pathogens, high prevalence disease agents, and normal or pathogenic flora that are reasonably likely to be encountered in a clinical sample. It is appropriate to conduct an in silico analysis of published genome sequences using the assay’s primers and probes. If in silico analyses of the target primers and probes reveal ≥ 80% homology between the cross-reactivity microorganism(s) and your test primers/ probe(s), we recommend that you conduct wet testing with that organism(s). We recommend using concentrations of 106 CFU/ml or higher for bacteria and 105 pfu/ml or higher for viruses. In silico analyses alone may be appropriate for organisms that are difficult to obtain. We recommend assessing potential cross-reactivity of the organisms listed in the table below, as applicable to the claimed sample type. If you are claiming an alternative matrix not listed below, please contact FDA to discuss the list of microorganisms recommended for testing.
[Please provide your complete cross-reactivity protocol, including a step-by-step description of how samples were prepared (e.g., starting concentration, dilution factor used to reach target concentration, volume of organism suspension, volume of clinical matrix, etc.) and tested with your device, the specific materials used to assess cross-reactivity and where these materials were obtained. Please include the Certificates of Analysis for each microorganism that is tested, or equivalent information (e.g., the culture protocol, lot number, manufacturing date, viral strain, a description of viral inactivation, pre-inactivation titer, and pre-inactivation sterility for viral isolates, etc.). For bacterial isolates, information may also include the isolate source, method for identification, number of passages, microbiological features, or other information. Please provide the line data for the cross-reactivity study as part of your request, in an Excel-compatible format. If the assay includes use of an analyzer or application to generate test results, please include the analyzer value with each test replicate.]
Recommended
List of Organisms to be Analyzed in
silico
and by Wet Testing for All Respiratory Samples
High priority pathogens from the same genetic family |
High priority organisms likely present in respiratory samples |
Human coronavirus 229E |
Adenovirus (e.g., C1 Ad. 71) |
Human coronavirus OC43 |
Human Metapneumovirus (hMPV) |
Human coronavirus HKU1 |
Parainfluenza virus 1-4 |
Human coronavirus NL63 |
Influenza A & B |
SARS-CoV-1 |
Enterovirus (e.g., EV68) |
MERS-coronavirus |
Respiratory syncytial virus |
|
Rhinovirus |
|
Chlamydia pneumoniae |
|
Haemophilus influenzae |
|
Legionella pneumophila |
|
Mycobacterium tuberculosis* |
|
Streptococcus pneumoniae |
|
Streptococcus pyogenes |
|
Bordetella pertussis |
|
Mycoplasma pneumoniae |
|
Pneumocystis jirovecii (PJP)* |
|
Pooled human nasal wash - to represent diverse microbial flora in the human respiratory tract |
|
Candida albicans |
|
Pseudomonas aeruginosa |
|
Staphylococcus epidermis |
|
Streptococcus salivarius |
*M. tuberculosis and P. jirovecii are applicable to lower respiratory matrices only (e.g., BAL, sputum, etc.).
High Priority Organisms Likely Present in Saliva Samples Recommended List of Organisms to be Analyzed in silico and by Wet Testing for Saliva Samples**
High priority pathogens from the same genetic family |
High priority organisms likely in the circulating area |
Human coronavirus 229E |
Adenovirus (e.g., C1 Ad. 71) |
Human coronavirus OC43 |
Human Metapneumovirus (hMPV) |
Human coronavirus HKU1 |
Parainfluenza virus 1-4 |
Human coronavirus NL63 |
Influenza A & B |
SARS-coronavirus |
Rhinovirus |
MERS-coronavirus |
Respiratory syncytial virus |
|
Herpes simplex virus type 1 (HSV-1) |
|
Epstein-Barr virus (EBV) |
|
Cytomegalovirus (CMV) |
|
Moraxella catarrhalis |
|
Porphyromonas gingivalis |
|
Bacteroides oralis |
|
Nocardia sp. |
|
Streptococcus mutans |
|
Streptococcus mitis or other Strep viridans |
|
Eikenella sp. |
|
Neisseria sp. |
|
Candida albicans |
|
Pseudomonas aeruginosa |
|
Staphylococcus epidermis |
|
Streptococcus salivarius |
|
Lactobacillus sp. |
**These organisms should be analyzed/tested in addition to the ones included in the immediately preceding table.
Microbial Interference Studies:
If in silico analysis reveals ≥ 80% homology between the cross-reactivity microorganisms and your test primers/ probe(s) set(s), we recommend that you either perform (1) a microbial interference study with SARS-CoV-2 and the microorganisms that your test primers/ probe(s) have homology to, or, as an alternative to the microbial interference study, (2) you may provide justification as to why (e.g., amount of primer(s)/ probe(s) included in your master mix) the performance of your test would not be impacted by the presence of a causative agent of a clinically significant co-infection, or (3) explain why the in silico results are clinically irrelevant (e.g., low prevalence of MERS-CoV, etc.).
Competitive microbial interference testing should be conducted for multiplex panels. The study should assess the effects of clinically relevant co-infections by testing selected microorganisms commonly found in the claimed sample matrix in the presence of SARS-CoV-2 at low concentration. The interference should be evaluated by testing with a minimum of 3 sample replicates spiked at a low (≤3x LoD) SARS-CoV-2 concentration and a high interferent level (preferably microorganisms), to represent the worst-case scenario. The interferent microorganisms can be tested individually or as a pool (of four or five) in the presence of low concentration of SARS-CoV-2. Each microorganism of a pool should be tested individually if that pool shows interference. If you plan to claim both upper and lower respiratory clinical samples, the study should be performed in the most challenging sample matrix, i.e., sputum. If interference is observed at the level tested, an additional titration study should be performed to determine the highest microorganism interferent level your test can achieve the stated performance.
[Please provide your complete microbial interference study protocol, including a step-by-step description of how samples were prepared (e.g., starting concentration, dilution factor used to reach target concentration, and volume of organism suspension for both inactivated SARS-CoV-2 and microbial interferent, volume of clinical matrix, etc.) and tested with your device, the specific materials used to assess microbial interference and where these materials were obtained. Please provide the line data for the microbial interference study in an Excel-compatible format. If the assay includes use of an analyzer or application to generate test results, please include the analyzer value with each test replicate.]
Endogenous/Exogenous Interference Substances Studies:
The extent of testing for studies of interference substances depends on the matrix that is indicated for the candidate test as well as on the technology of the candidate test. If the candidate test uses extraction methods not previously reviewed by FDA as part of premarket submission or the candidate test does not use an extraction procedure (as for example, many point-of-care tests), we recommend testing for potential interferents. The following Table includes suggested potential interferents that might be appropriate to test for a test indicated for upper respiratory samples and/or oral fluid or saliva. We recommend testing the following substances listed in the table below, as applicable based on the indicated matrices, with and without inactivated virus at 2-3x LoD in three replicates for each substance. Please contact FDA if you have questions about appropriate study designs.
List of Potential Interfering Substances Recommended for Testing When the Candidate Test is Indicated for Respiratory Samples
-
Potential Interfering Substances
Concentration
Afrin Original nasal spray
15% v/v
Sore throat and cough lozenges such as Cepacol Lozenges (benzocaine/menthol)
3 mg/mL
Chloroseptic Sore Throat spray
5% v/v
Mouth Wash (Saliva)
5% v/v
Cough syrup (e.g., Robitussin)
5%
Mucin: bovine submaxillary gland, type I-S
2.5 mg/ml
Nicotine or Tobacco
0.03 mg/ml
Toothpaste (Saliva)
0.5% v/v
[If a concentration is not listed in the table above, please determine an appropriate concentration and provide the scientific justification supporting your proposed concentration as part of your EUA request.]
Testing should be conducted to demonstrate sample stability throughout the real-world conditions in which they are collected and tested, according to your instructions for use. When the test is intended to be performed on the sample immediately or shortly after obtaining the sample, sample stability testing could be relatively short (i.e., 2 hours at room temperature) and conducted with contrived samples at 3x LoD using inactivated virus spiked into negative clinical matrix. If you intend to test retrospective clinical samples that have been frozen, you should also conduct fresh versus frozen studies to support use of these samples.
LoD Target Level |
Number of Samples |
3-5 times LoD |
10 |
1-2 times LoD |
30 |
Negative |
10 |
Total |
50 |
[Please provide a complete sample stability protocol, including a detailed, step-by-step description of how you prepared and tested each replicate, and provide all study data in an Excel-compatible format, with analyzer values, if applicable. The protocol should also include sample stability information, including the study design and results if the sample is shipped to a testing site from another location (e.g., samples collected at home or physician’s office).]
FDA recommends conducting prospective, blinded, randomized clinical agreement trials with at least 30 positive samples and 30 negative natural clinical samples (prospective, retrospective, or leftover) from patients suspected of COVID-19 by their healthcare provider. The number of negative samples may vary according to the disease prevalence at the time of your study. Evaluations with contrived clinical samples are inadequate to support the clinical performance of molecular diagnostic tests at this time.
If you seek authorization for multiple sample types, each sample type should be evaluated. This may be done by collecting samples from different anatomical sites from the same patient. To minimize the occurrence of discordant results due to biological variability, both samples should be collected within a short time period (e.g., within the same healthcare visit). Types of upper and lower respiratory samples are noted by the CDC at the following website: https://www.cdc.gov/coronavirus/2019-ncov/lab/guidelines-clinical-specimens.html.19 Please note that specimens with and without VTM are considered two distinct types of specimens. If you are seeking indications for testing with sputum and any other respiratory sample, we recommend testing either 30 sputum samples or a combination of upper respiratory samples and sputum samples, such as 15 NP and 15 sputum samples, or 15 combined upper respiratory samples and 15 sputum samples.
You may use frozen samples if you demonstrate analytically that preservation of samples (e.g., by freezing at ≤-70°C) does not affect the accuracy of test results compared to freshly collected samples.
You may use samples that previously tested positive by another authorized RT-PCR assay without additional comparator testing. [Please provide the type and source of the samples, results, and numerical output signals such as Ct values or numerical output for each tested sample, and the initial test date.]
Approximately 25% of the positive samples should have a low viral load (i.e., low positives) as measured by the comparator test (i.e., Ct values should be within 3 Ct of the mean Ct at the LoD of the comparator test). When conducting a prospective study, if fewer than 25% of positive samples are low positives per the comparator assay, the prospective samples may be supplemented with additional low positive samples (i.e., archived samples, samples collected from convalescent patients, etc.).
Samples from each individual should be evaluated with the candidate test and an authorized RT-PCR test which uses a chemical lysis step followed by solid phase extraction of nucleic acid (e.g., silica bead extraction) and reports a Ct value. The comparator test may have the same, or different, targets as the candidate test; however, different primer and probe regions are recommended. The comparator test should be one of the more sensitive RT-PCR assays authorized by FDA. We encourage you to review the results from the FDA SARS-CoV-2 Reference Panel20 and contact us to discuss your choice of comparator test. Evaluations with the comparator test should be conducted per the authorized instructions for use. If any modifications are made to the authorized comparator test, additional bridging studies may be necessary. Please contact FDA if you are considering using a modified configuration of an authorized RT-PCR assay.
When collecting samples, the standard of care sample (i.e., the sample used for clinical and not investigational purposes) should always be collected first, including when the comparator test is also the standard of care. If the comparator test is not the standard of care, swabs taken from the same anatomical area for the comparator test and candidate test (e.g., anterior nasal swabs, oropharyngeal (OP) swabs, etc.) should be randomized to ensure that bias is not introduced due to an unequal distribution of viral materials. When two distinct anatomical sites are being assessed, it is not necessary to randomize sample collection order (e.g., saliva compared to NP swabs).
You may consider use of an enrichment strategy in which individuals with a known COVID-19 infection status are invited to participate in your clinical evaluation study. If using an enrichment strategy, you should carefully consider how you will randomize and blind operators to the participant’s infection status and minimize potential bias. Data from an enriched study design should represent the full range of viral loads, with both low and high positives samples. Please contact FDA to discuss any alternative study designs or enrichment strategies.
All clinical samples tested in your study should be evaluated in accordance with the candidate test’s proposed diagnostic algorithm (i.e., tested using the procedure in the instructions for use), including retesting when appropriate. The limited volume of natural samples may preclude retesting. In instances where retesting is indicated but not performed, for the purposes of performance evaluation, initial results should be analyzed for performance and equivocal/indeterminate/inconclusive results should count against your final performance. Samples should be tested in a blinded fashion, e.g., positive, and negative samples should be presented to the end user in a blinded fashion. The end user should also be blinded to the results of any comparator method testing.
FDA recommends establishing a discordant analysis plan prior to your clinical study. Discordant samples should be tested with a second EUA authorized RT-PCR test that has also demonstrated high sensitivity, and which uses a chemical lysis step followed by solid phase extraction of nucleic acids (e.g., silica bead extraction). Results from a Discrepant analysis should not be included in the calculation of negative percent agreement (NPA) and positive percent agreement (PPA) but may be added to the performance table as a footnote.
Studies involving clinical samples (human specimens) conducted in support of an EUA request are subject to applicable requirements for Institutional Review Board (IRB) review and approval and informed consent (see 21 CFR parts 50, 56, and 812). FDA’s policy regarding informed consent requirements for certain studies using leftover, de-identified samples is outlined in the FDA guidance “Guidance on Informed Consent for In Vitro Diagnostic Device Studies Using Leftover Human Specimens that are Not Individually Identifiable.”21
Candidate tests should demonstrate a minimum of 95% positive and negative agreement for all sample types requested. In addition, for studies where multiple sample types from the same patient are evaluated by the candidate test, there should be no significant evidence of a trend in Ct values that is indicative of the potential for false negative results.
[Please
describe the clinical study used to evaluate the clinical performance
of the test.]
[Please specify how the samples were generated, collected, and sourced. Please also specify if the samples were fully prospective or a mix of prospective and retrospective. Please specify inclusion/exclusion criteria, collection and testing sites, number of samples collected and tested, and number of operators performing the testing.]
Clinical Evaluation for Screening Individuals Without Symptoms or Other Reasons to Suspect COVID-19:
The recommendations below reflect FDA’s current thinking. The study design and recommendations may change as additional information becomes available regarding asymptomatic infections, including but not limited to viral titer dynamics and transmission rates in this population.
If you seek claims for screening individuals without symptoms or other reasons to suspect COVID-19, FDA recommends that you conduct a prospective clinical study in asymptomatic individuals free of any symptoms of SARS-CoV-2 infection for at least two weeks prior to enrollment and testing, if not known to be previously positive. Asymptomatic individuals who are suspected of COVID-19 (i.e., via exposure) should be excluded. As part of your clinical study protocol and data, you should document how you screened and confirmed that all enrolled individuals were asymptomatic and consistent with your proposed intended use.
You should follow the clinical study recommendations listed above (i.e., 30 symptomatic positive and negative samples), except that the number of enrolled patients should be sufficient to ensure at least 20 positive samples (as per a highly sensitive comparator test) are consecutively collected. A minimum of 100 consecutive SARS-CoV-2 negative samples (as per the comparator test), should be collected. The total number of samples needed will depend on the prevalence of SARS-CoV-2 in the intended use population. If less than 20 SARS-CoV-2 positive samples, as determined by the highly sensitive comparator test, are obtained from the prospective study, or prevalence in the test population is very low, you may consider enrichment strategies. For example, you may conduct an additional prospective study in an asymptomatic screening population that is under quarantine due to possible exposure, to increase the chances of obtaining more SARS-CoV-2 positive samples. Please consult FDA prior to implementing enrichment approaches in your clinical study design.
In the clinical study, you should compare results from your candidate test and a highly sensitive comparator test for each patient enrolled. Please refer to recommendations above regarding selection of a comparator test. Samples for the candidate test should be collected according to the instruction for use. Samples for testing with the comparator test should be healthcare provider collected NP swabs. If an NP swab cannot be collected, a MT swab may be used. Sampling for the candidate test and comparator test should occur within a short timeframe to avoid biological variability in viral load.
In the context of an EUA request, FDA considers samples collected at one or two sites to be appropriate. However, if you are planning to use the same data to support a subsequent De Novo/510(k) submission, we recommend that you collect samples at a minimum of three geographically and demographically diverse sites.
It may be possible to use archived samples that were collected from asymptomatic patients. We recommend you contact FDA to discuss such an approach prior to initiating your study.
The data should be sufficient to demonstrate the following minimum performance:
PPA ≥95% (Lower Bound of the two-sided 95% confidence interval >76%)
NPA ≥98% (Lower Bound of the two-sided 95% confidence interval >95%)
Adding Population Screening of Individuals Without Symptoms or Other Reasons to Suspect COVID-19 to an Authorized Test:
Alternative approaches may be appropriate for candidate tests that have been previously authorized for use with common upper respiratory samples (i.e., NP, MT, nasal swabs, etc.) from symptomatic patients. For example, if your assay is highly sensitive as determined by testing with the FDA SARS-CoV-2 Reference Panel or a recognized international standard, FDA will consider expanding your authorization to cover asymptomatic screening with a condition of authorization that you complete the study outlined above. Developers may also refer to the Supplemental Template for Developers of Molecular and Antigen Diagnostic COVID-19 Tests for Screening with Serial Testing22 for additional options when seeking a screening claim with serial testing when a clinical evaluation with symptomatic patients suspected of COVID-19 infection by their healthcare providers has been performed. FDA is open to considering additional alternative study designs to demonstrate that the performance of the candidate test is appropriate for screening individuals without symptoms or other reasons to suspect COVID-19. We recommend contacting FDA to discuss alternative study designs prior to beginning such a study.
Combining multiple patient samples to create one pooled sample for testing could enable broader access to testing by increasing throughput, though it may also reduce the sensitivity of a test because samples are diluted. Therefore, FDA does not recommend sample pooling for tests with performance <95% PPA when testing individual samples, based on validation with positive patient samples. Use of pooling should be considered in the context of the positivity rate of a test in the test population, the analytical sensitivity of the test, and the percent of weak positive subjects in the tested population. The impact of decreased analytical sensitivity depends on the percent of subject samples with viral genetic material concentrations close to the LoD (weak positives) in the tested population. Therefore, analytical sensitivity of the test with n-sample pools should be evaluated, where n is the number of samples included in the pool. When resource availability is sufficient to meet testing demand, FDA recommends considering whether the risks of reduced test sensitivity with pooling continue to outweigh the benefits of resource conservation.
When pooling, a negative result implies that all samples in the pool are negative. A positive result indicates that at least one sample in the pool is positive. When an n-sample pool is positive, each sample within the pool must be individually tested to determine which is/are positive. Due to the reduction in analytical sensitivity, the test report should state that pooling was used during testing.
A test validated for a specific n-sample pooling strategy is also considered to be validated for any number of pooled samples below n. For example, a test validated for a 5-sample pooling strategy can be performed for any n≤5 pools.
If you seek authorization for testing of pooled specimens, your instructions for use should include instructions for laboratories to select and implement an initial validated sample pool size and to perform ongoing monitoring of the selected and implemented pool size.
Pooling Implementation – Determine the appropriate pool size based on percent positivity rate in the testing population and pooled testing efficiency. For swab pooling, this should include a detailed procedure describing a method to combine swabs into a single volume of transport media. The procedure should include recommendations to maximize the amount of sample resuspended into the transport media from the swab and help ensure that the user performs sample and swab handling in a manner consistent with current infection control procedures, which should also reduce the chance of carryover between sample pools.
Pooling Monitoring – Monitor the positivity rate from pooled samples.
Pooling Re-assessment – Reassess the impact of pooled testing on test performance.
These activities are described in more detail in Appendix B.
FDA is providing recommendations for two approaches to patient sample pooling: 1) pooling aliquots of transport media which each contain a single patient sample (media pooling) or 2) adding swabs from multiple patients into a single volume of transport media (swab pooling).
When pooling transport media, one individual sample is defined as a single sample swab collected from a subject and placed in a specific volume of transport media. In this type of pooling, an aliquot of each individual sample is combined into non-overlapping pools of n samples and each n-sample pool is tested. Therefore, the instructions for use should specify a sample volume great enough to allow for individual and pooled testing so that, during clinical use, any samples in a positive pool can be re-tested without the need for a second sample collection.
FDA believes an n=5 is a reasonable starting point for validation of pooling for a high-sensitivity test in populations with a positivity rate of approximately 5% to 6%. In populations with a lower positivity rate, larger sample pools may be feasible. In populations with higher prevalence, smaller sample pools may be needed. FDA recommends that developers begin by validating their tests for pooling using an n=5. Tests validated and authorized for n=5 can then be used with any n≤5 pools, depending on testing needs and taking into consideration local positivity rate.
We strongly recommend that test developers develop and validate a system for deconvoluting pooled test data which is intended to accurately identify individual patient samples composing each pooled sample. If a test developer plans to use a software solution intended to deconvolute pooled SARS-CoV-2 diagnostic test data, then we recommend providing validation data establishing that the software can achieve its intended use. For example, we recommend including evidence that the software has been validated to ensure that:
The inputs and outputs of the software are appropriate for the intended use of the candidate test;
All expected inputs produce the expected outputs for all functions critical for system operation; and
The system will be provided to the customer free of defects or defects will be known and mitigated.
Please see section I(4) for more information on appropriate software validation approaches.
To add an n-sample pooling strategy to an authorized assay, you should submit a supplemental EUA request with the appropriate validation data, as described in Appendix A. If the authorized assay has a PPA ≥ 95% when testing individual samples, based on validation with positive patient samples, the authorized assay may be used as the comparator test for the pooling validation study.
To include an n-sample pooling strategy for a candidate test that has not been previously authorized, you should submit an EUA request with the appropriate validation data for individual testing in the proposed intended use population (see section J(5)) as well as appropriate validation data for sample pooling, as described in Appendix A.
Swab pooling is an approach which conserves transport media and has the potential to maintain sensitivity of the test; however, deconvoluting which swab was positive cannot be done without collecting another sample. This approach also results in a high concentration of swab samples in transport media and thus inhibition may be observed. The effects of inhibition due to high concentrations of swab samples (e.g., mucin) and high concentrations of virus when there are multiple positive swabs in the swab pool should be investigated.
The validation recommendations for swab pooling are the same for tests that have and have not been previously authorized for individual sample testing. We recommend performing the two swab pooling validation studies using the highest number of swabs that is both desired and deemed feasible. If the data do not meet the acceptance criteria noted below, we recommend evaluating a lower number of swabs until the recommended acceptance criteria are met. Laboratories can proceed with testing with any number of pooled swabs up to the highest number of pooled swabs that was successfully validated.
If the candidate test has not been previously authorized for individual sample testing, test developers should submit an EUA request that also includes the appropriate validation for individual sample testing in the proposed intended use population. Refer to section J(5) of this template for recommendations regarding recommended clinical evaluation of individual sample testing.
Swab pooling validation:
[For n-swab pooling strategies, the two studies below should be conducted, and the results included in your EUA request]:
You should establish performance related to test interference from multiple swab samples in a single volume of transport media. N-swab samples containing the maximum number of swabs you intend to validate in the minimum volume of transport media you intend to validate should be tested with an analyte concentration of 2-3X LoD for the individual swab. The swabs should contain clinical matrix negative for SARS-CoV-2. The acceptable range of transport media volume and the maximum number of swabs should be noted in your instructions for use. We recommend testing replicates of three n-swab pooling samples at the same analyte concentration both with and without SARS-CoV-2.
For example, if the instructions for use for the candidate test recommends pooling three swabs (n = 3), then we recommend acquiring a total of nine confirmed negative swabs from individual subjects and adding three unique swabs to three unique tubes of transport media, thereby making three n-swab pooling samples. Each n-swab pooling sample should be spiked with either a positive patient sample (in transport media), live virus, or inactivated virus at a concentration of 2-3X the LoD of the candidate test. We recommend testing a total of at least 20 replicates, which can be composed of equal numbers of aliquots taken from each n-swab pooling sample (i.e., 7 replicates from each sample in this example). Ideally, negative n-swab sample matrix should be tested prior to spiking to ensure that the matrix is negative. Acceptance criteria should be at least 95% agreement with the expected results and an invalid rate of < 5%. [Please include the Ct value line data (if applicable) in your EUA request.]
You should evaluate the effect of high viral concentrations on candidate test performance. It appears that patients with SARS-CoV-2 infection can exhibit unusually high viral loads. This, combined with the possibility of pooling multiple positive swabs into a single volume of transport media, could result in unexpectedly high viral titer in the pooled sample. We recommend evaluating existing data on viral loads in infected subjects and, in combination with your existing LoD data, propose a maximum expected viral titer per swab. Using this number, estimate the expected viral titer in transport media with at least three positive swabs. For instance, if you expect a maximum of 100,000X LoD per swab, we recommend spiking a single negative n-swab sample with 300,000X LoD target analyte and testing with 10 replicates. It is anticipated that all replicates are either positive or have an invalid rate of ≤5%.
[If the device is intended for POC testing, please provide a detailed study description and data to demonstrate that non-laboratory healthcare providers can perform the test accurately in the intended use environment]. Your studies to support a POC claim should include the following: (1) a POC clinical evaluation including use of appropriate sites and test users, (2) supplemental POC samples, and (3) POC flex studies. For more details, please see each section below
The clinical study design should reflect how the test will be used in clinical practice. It is expected that a test with “POC” designation will be widely used in CLIA waived medical facilities (e.g., physician office, outpatient clinic, emergency room (ER)), but also in less traditional settings (e.g., tents, schools, etc., with health care provider oversight of testing) where health care providers are present.
You should select one or two non-laboratory sites in the United States (U.S.) to assure that the operators are representative of intended operators in the U.S., e.g., doctor’s office, ER, outpatient clinic, drive-through testing facility, or another area in a medical facility outside the central laboratory where samples are collected and tested in real time. This would allow evaluation of the sample collection and handling, including addition into the sample port/well of the test, both of which may be significant sources of error. Four to six operators, representing intended healthcare provider operators, but who are not laboratory trained (e.g., nurses, nursing assistants and doctors) should participate in the study. Testing should be performed using only Quick Reference Instructions (QRI) - supplemental materials, such as a video or a mobile application that can be easily accessed by the user, are encouraged to be included with the proposed candidate test but should not be used during this study to mimic the worst-case scenario.
[Please provide the detailed individual replicate result data in an Excel-compatible formant and protocols for each of your studies, including:
The objective of the study;
Detailed test procedure;
Materials used;
A list of samples tested;
Results (presented in tabular format), including invalid results;
Conclusions;
Any appropriate mitigation measures (e.g., labeling changes, changes to test design, etc.); and
Operator background (e.g., education, training, experience, etc.)
As part of your EUA request, please include a table in which your study results are stratified by operator.]
A description of an appropriate clinical comparator test is included in section J7 above.
A total of 30 prospectively collected positive (confirmed by an EUA-authorized test) and 30 negative natural clinical samples should be tested (mock clinical samples are not appropriate). Testing should be conducted for at least 2 weeks. If an insufficient number of positive results is observed after such time (<30), you may collect samples at another site to ship to the testing site or use banked samples to supplement your positive samples. Banked samples should not be pre-selected based on Ct value and should be presented blinded (mixed with negatives) to the testing site. Ideally, the same comparator test should be used for banked and prospectively collected samples.
Clinical Performance
A molecular POC candidate test should demonstrate positive and negative agreement of ≥ 95%. However positive agreement of ≥ 80% may be considered with appropriate limitations added to the intended use that would mitigate the risk of false negative results. For example, negative results may be considered presumptive negative if the demonstrated PPA is lower than 95%.
You should also conduct testing with samples prepared with SARS-CoV-2 viral load near the LoD of your assay in clinical matrix. The testing should be conducted by minimally trained operators and should consist of 10 low positives (<2 times LoD) and 10 negative samples per site. All contrived samples should be blinded and randomized and each operator should test at least three low positive and three negative samples integrated into the site’s workflow with the clinical samples above. These samples are intended to supplement, not replace, the clinical samples in your study.
[Please include a table in an Excel-compatible format in which your study results are stratified by operator.]
You should also conduct a thorough hazard analysis considering the main known sources of errors. Based upon your hazard analysis, you should conduct flex studies to evaluate the impact of errors, or out-of-specifications conditions, on the candidate test performance. Each sample should be prepared at 2xLoD in negative clinical matrix and should be evaluated in three replicates for each condition under evaluation. Flex studies can be conducted with trained operators at an internal testing site. Each study should be performed using a pre-defined study protocol that includes the following:
The objective of the study;
Detailed test procedure; and
Materials used.
Potential stress conditions include:
40°C and 95% room humidity (RH) (mimicking hot and humid climates);
Delay in sample testing or reading time;
Delay and/or disturbance in operational steps;
Sample volume variability;
Buffer volume variability;
Read time variability; and
Other, as appropriate.
[Please provide a detailed, step-by-step description of how you prepared and tested each replicate and provide all study data in an Excel compatible format, with analyzer values, if applicable. Data for each sample evaluated (i.e., line data) should be provided. If erroneous results are observed during studies evaluating the robustness of the device, adequate mitigation(s) should be provided.]
Please see the Template for Developers of Molecular and Antigen Diagnostic COVID-19 Tests for Home Use23 for more in-depth flex study designs. Alternative sources of information on flex studies that may be appropriate for the candidate test can be found on the FDA CDRH website containing CLIA Waiver by Application Decision Summaries.24
If you are requesting an EUA for a multi-analyte respiratory panel, analytical and clinical evaluations for each target analyte should be included. We recommend considering the study designs and data summaries noted in the published EUA Summaries and Instructions For Use of authorized multi-analyte tests.
If you are planning to use the Right of Reference to the CDC Influenza SARS-CoV-2 (Flu SC2) performance data, please see the web page https://www.cdc.gov/coronavirus/2019-ncov/lab/multiplex-faq.html25 for more information.
To add the SARS-CoV-2 target to respiratory panels previously cleared by the FDA where the SARS-CoV-2 reagents are run in a separate well (or tube) and no modifications are required to the cleared portion of the panel, only studies for validation of the SARS-CoV-2 reagents previously described in this template are recommended.
To add the SARS-CoV-2 target to respiratory panels previously cleared by the FDA where the SARS-CoV-2 reagents are combined in the same well as the reagents for previously cleared analytes (in a multiplex reaction), the following studies should be conducted to validate the SARS-CoV-2 reagents and the modifications made to the cleared respiratory panel:
Studies previously described in this template to validate the SARS-CoV-2 reagents
LoD confirmation of the previously cleared analytes by conducting side by side testing of 3-5 replicates of serially diluted viruses with modified and original versions of the test to show that the LoD is unchanged due to modifications
Testing 10 retrospective positive samples for each previously cleared analyte
Competitive inhibition study with clinically relevant titers of each analyte in the panel (viruses 105 PFU/mL, bacteria 106 CFU/mL)
To support an EUA for a multi-analyte respiratory panel candidate test that was not previously cleared by FDA, analytical and clinical evaluations for each target analyte should be provided.
A. Analytical Performance:
The following analytical studies should be conducted by wet testing and data provided to the FDA for review:
Limit of Detection (Analytical Sensitivity)
Cross-Reactivity / Microbial Interference
Inclusivity / Analytical Reactivity
Collection Media Equivalency – each claimed additional sample collection media not used in your clinical study should be validated (if appropriate for study designs)
Competitive microbial interference (for organisms for which the test is not indicated for use)
Competitive inhibition (for analytes for which the test is indicated for use)
Interfering Substances Study (Endogenous and Exogenous)
Clinical Sample Stability
Reagent Stability testing protocol
Carry over/Cross-Contamination (if a new instrument not previously cleared/approved by the FDA is used with the candidate test)
Reproducibility and Repeatability (if a new instrument not previously cleared/approved by the FDA is used with the candidate test)
Fresh vs. Frozen Samples. If you intend submit data testing archived frozen samples in support of your EUA request, please conduct an analytical study to demonstrate that preservation of samples (e.g., by freezing at ≤-70°C) does not affect the accuracy of test results compared to freshly collected samples.
To evaluate the clinical performance of your multi-analyte candidate test, a prospective clinical study should be conducted. Considering the public health needs in the current emergency, a clinical performance study in support of the EUA request may be conducted at one site testing archived positive and negative clinical samples with known sample types. The pre-selection of archived positive samples should represent a range of viral load or Ct values including low positive samples near the candidate test cut-off.
For the non-SARS-CoV-2 analytes, such as Influenza A, Influenza B, and Respiratory Syncytial Virus (RSV), etc., a minimum of 50 positive Influenza A, 30 positive Influenza B, and 30 positive RSV archived samples should be included in the clinical study.
Since your candidate test has not been FDA-cleared for the respiratory pathogens for which it is indicated, and it is likely that the candidate test would be used in patients with respiratory symptoms in lieu of an FDA-cleared respiratory panel, FDA generally intends to include a condition of authorization that you conduct a post EUA prospective clinical study. The prospective clinical study should include a minimum of three sample collection sites and three testing sites, prospectively enrolling patients with general respiratory symptoms. Until the post-EUA prospective clinical study is completed, and the study results are reviewed by the FDA, FDA recommends including a warning/limiting statement in the instructions for use for your test indicating that results (positive and negative) for the non-SARS-CoV-2 analytes should be confirmed with an FDA-cleared nucleic acid amplification test (NAAT) if clinically indicated.
For multiplex candidate devices that detect and differentiate SARS-CoV-2, Influenza A/B, and RSV viral nucleic acids, a minimum of 50 positive SARS-CoV-2, 50 positive Influenza A, 30 positive Influenza B, and 30 positive RSV prospectively collected and tested samples should be included in the prospective clinical study enrolling prospective samples (an all-comers study) post-authorization.
The clinical performance of the candidate test for the non-SARS-CoV-2 analytes (e.g., Influenza A/B and RSV, etc.) should be determined by comparison to an FDA-cleared molecular test used as a comparator test. Using an FDA-cleared molecular test with prospective clinical study data from the past 5 years as the comparator test for assessing clinical performance of your device is recommended.
Since most FDA-cleared comparator test options have been validated and cleared for use with NP swab and/or nasal swab samples only, if you intend to assess clinical performance of your device testing other typical upper respiratory tract sample types (e.g., nasal mid-turbinate swab, nasopharyngeal wash/aspirate, nasal wash/aspirate, and oropharyngeal swabs, etc.), and the FDA-cleared molecular test you intend to use as the comparator test has not been cleared for use with the other typical upper respiratory tract sample types you wish to claim, you should conduct a paired-sample study in which one of the paired samples of an FDA-cleared sample type (e.g., NPS or NS) is tested with the FDA-cleared comparator test and the other paired sample of a typical upper respiratory tract sample type you intend to claim (e.g., nasal mid-turbinate swab or nasopharyngeal wash, etc.) is tested with your candidate device. Alternatively, as a less burdensome approach, you could validate the FDA-cleared comparator test for use with the desired sample type by performing an analytical LoD comparison study between the desired typical upper respiratory tract sample type and an FDA-cleared upper respiratory tract sample type using the comparator test prior to initiating the clinical evaluations.
If you intend to assess clinical performance of your device testing an atypical sample type (e.g., saliva, buccal swabs, etc.), and the FDA-cleared molecular test you intend to use as the comparator test has not been cleared for use with the atypical sample type you wish to claim, you should conduct a paired-sample study in which one of the paired samples of an FDA-cleared sample type (e.g., NPS or NS) is tested with the FDA-cleared comparator test and the other paired sample of an atypical sample type you intend to claim (e.g., saliva, oral fluid, and buccal swabs, etc.) is tested with your candidate device. The alternative approach described above for the typical upper respiratory tract sample types is not appropriate, and therefore, not appropriate, for the atypical sample types.
The performance expectation and recommended comparator method for SARS-CoV-2 are noted in Section J(5). For Flu A/B, and other respiratory viruses, PPA should be >90% (with a lower bound of the two-sided 95% confidence interval >80%), and the NPA should be >95% (with a lower bound of the two-sided 95% CI >90%) in comparison to an FDA-cleared molecular test.
We recommend that you submit a Pre-EUA with an outline of the studies that you plan to conduct to support the FDA-authorization or contact FDA at CDRH-EUA-Templates@fda.hhs.gov for specific feedback.
Claiming Multiple Typical Upper Respiratory Tract Sample Types for use with Multi-analyte Panels not Previously Cleared by the FDA:
We recommend that you conduct an LoD study using the most challenging typical upper respiratory tract sample type you wish to claim with your candidate device (e.g., NP swab and nasal swab, etc.). In addition, you should attempt to include all typical upper respiratory tract sample types that you intend to claim for use with the candidate test in the pre-authorization clinical performance evaluation study. If you are unable to include all desired typical upper respiratory tract sample types in the pre-authorization clinical performance evaluation study, after demonstrating due diligence on your part to enroll such samples, you may claim the following typical upper respiratory tract sample types if you have validated your device analytically and clinically with NP swabs, nasal swabs, and/or MT swabs: NP swabs, anterior nasal swabs, MT swabs, NP washes/aspirates, nasal washes/aspirates, and oropharyngeal swabs, as long as all your claimed typical upper respiratory tract sample types are incorporated into the post-authorization prospective clinical study. For any typical upper respiratory tract sample type(s) that was/were not evaluated in the pre-authorization clinical study, we recommend including a limiting statement in your test’s labeling indicating that the performance of your test testing such typical upper respiratory tract sample type(s) has not been evaluated. Once the post-authorization prospective clinical study is completed and the clinical performance of your test testing such typical upper respiratory tract sample type(s) is deemed appropriate by the FDA, you may remove this limiting statement from your test’s labeling.
Atypical sample types (e.g., saliva, oral fluid, and buccal swabs, etc.) and lower respiratory tract sample types (e.g., BAL and sputum, etc.) should be validated with LoD studies performed in each of the claimed sample matrices. Additionally, both the pre-authorization and post-authorization clinical evaluation studies should include these sample types in the study design.
FDA recommends the following analytical and clinical validation to validate use of a new test with multiple thermocyclers and extraction methods.
Limit of Detection (LoD):
These studies should be repeated for each clinical matrix for which the candidate test is indicated for use. Pick one RT-PCR instrument and determine the tentative LoD (using 5 replicates in 10-fold dilution) followed by the confirmatory LoD (20 replicates spiked at tentative LoD) for each extraction method on the chosen instrument. Note: If you detect 20/20 replicates in your confirmatory LOD study you should test the next lower concentration, using a 3-fold dilution, until you achieve a hit rate of <20/20.
If the different extraction methods yield a similar LoD (≤3 times LOD) on the RT-PCR instrument chosen for initial testing, pick one extraction method for further LoD determination on the remaining RT-PCR instruments and follow the recommendations below.
If the extraction methods do not yield a similar LoD on the chosen RT-PCR instrument, please choose the extraction method with the worst LoD for further comparison of the LoD on all RT-PCR instruments.
For all other RT-PCR instruments you should use the following adaptive LoD study design:
Please perform a refined tentative LoD study with 5 replicates at 0.5, 1, and 1.5 to 2 times LoD. If you detect 4/5 replicates as positive at all the tested levels, you need to include the next higher concentration (i.e., 3 times LoD). If you obtain 5/5 replicates at 0.5 times LoD, you need to test the next lower concentration (i.e., 0.25 times LoD). You should test in this manner until you find the lowest concentration that gives you 5/5 positive results for the tested RT-PCR instrument. This concentration should be used for a confirmatory LoD study for that RT-PCR instrument using 20 replicates.
Final reported LoD: [Please include in your EUA request a list of all RT-PCR instruments with their respective LoDs, if different LoDs are obtained.] LoDs are considered comparable if they are between 1-3 times LoD. These studies should be repeated for each clinical matrix for which the candidate test is indicated.
Note, if there are differences in the extraction input volume, extraction elution volume, and PCR input volume (extracted nucleic acid) then the LoD should be confirmed for each.
Interference Substances Studies (if applicable):
FDA recommends evaluating interfering substances with the extraction method and RT-PCR instrument combination that has the least sensitive LoD.
Inclusivity Testing:
FDA recommends evaluating inclusivity with the extraction method and RT-PCR instrument combination that has the least sensitive overall LoD.
Exclusivity Testing:
FDA recommends evaluating exclusivity with all extraction/instrument combinations.
Clinical study:
If an LoD study confirms equivalency for all RT-PCR instruments (between 2-3 times LoD), then the clinical study may be conducted with any RT-PCR instrument. If one or more RT-PCR instruments have different LoDs, we recommend conducting the clinical study with the extraction method / RT-PCR instrument combination with the worst LoD.
K. UNMET NEED ADDRESSED BY THE PRODUCT
This section will be completed by FDA.
L. APPROVED/CLEARED ALTERNATIVE PRODUCTS
There is no adequate, approved, and available alternative to the emergency use of the product.
This section will be completed by FDA.
N. FACT SHEET FOR HEALTHCARE PROVIDERS AND PATIENTS:
During review, FDA will make available Fact Sheet templates. See examples for authorized tests on our website.26
O. INSTRUCTIONS FOR USE/ PROPOSED LABELING/PACKAGE INSERT:
[You should include Instructions for Use, Box Labels, Vial Labels, and any other proposed labeling.]
P. RECORD KEEPING AND REPORTING INFORMATION TO FDA:
As allowed by Section 564(e) of the FD&C Act, FDA may require certain conditions as part of an EUA. FDA generally includes the following record keeping and reporting information requirements in the EUA.
[Test Developer name] will track adverse events and report to FDA under 21 CFR Part 803. A website is available to report on adverse events, and this website27 is referenced in the Fact Sheet for Health Care providers as well as through the [Test Developer name] Product Support website: [Include link to Test Developer’s Website]. Each report of an adverse event will be processed according to [Test Developer name]’s Non-Conformance Reporting Requirements, and Medical Device Reports will be filed with the FDA as required. Through a process of inventory control, [Test Developer name] will also maintain records of device usage/purchase. [Test Developer name] will collect information on the performance of the test, and report to FDA any suspected occurrence of false positive or false negative results of which [Test Developer name] becomes aware. [Test Developer name] will maintain records associated with this EUA and ensure these records are maintained until notified by FDA. Such records will be made available to FDA for inspection upon request.
Appendix A: Media Pooling Validation
Preliminary Clinical Sample Pooling Validation Study
The test developer should conduct a preliminary clinical sample pooling validation study with individual positive clinical samples, comparing the performance of the candidate test when testing n-sample pools to the performance of a comparator test when assaying individual samples. We recommend using only a high sensitivity EUA-authorized RT-PCR assay which uses a chemical lysis step followed by solid phase extraction of nucleic acid (e.g., silica bead extraction) as the comparator test. If available, FDA recommends selecting a comparator test that has established high sensitivity with an internationally recognized standard or FDA SARS-CoV-2 Reference Panel. 28 This study design is written assuming the study uses a separate comparator test. As discussed earlier, if requesting to add pooling to a previously authorized test, a separate comparator may not be needed, and this study design can be modified accordingly.
We recommend that the test developer evaluate in this preliminary clinical sample pooling validation study a minimum of 20 positive clinical samples (can be archived samples), as determined by the comparator test, with sufficient volume. The 20 samples should include 25% weak positive samples, as determined by the comparator test (i.e., (Ct values in a SARS-CoV-2 positive samples should be within 1-3 Ct of the mean Ct at the LoD of the comparator test). If weak positive samples, as determined by the comparator test, represent less than 25% of the total number of positive samples, the test developer should preferably supplement the validation sample set with additional natural weak positive samples to make up to 25% of the total enrolled positive samples. However, diluting individual natural positive samples with individual or pooled natural negative samples to reach the 25% goal is also appropriate. If archived samples are acquired, the test developer should consider using the most recently archived samples to minimize the potential risk of sample degradation due to prolonged storage.
All positive samples in the preliminary clinical sample pooling validation study should be individually tested by the candidate test and the comparator test. To characterize the performance of the candidate test when testing pooled samples, each individual positive sample (as determined by the comparator test) should be pooled with n-1 (e.g., where n=5, n-1=4) negative samples, as determined by the candidate test. The resulting sample pools, each consisting of 1 positive sample and n-1 negative samples, should be tested by the candidate test.
In order to construct the 20 n-sample pools for testing in this preliminary validation study, the test developer should use an appropriate number of individual negative samples (as determined by the candidate test) from the intended use population. While the (n-1) negative samples in a given n-sample positive pool should all be different negative samples collected from unique individuals, the same negative samples may be used in building different n-sample positive pools.
Based on this recommended study design, the preliminary clinical sample pooling validation study should generate the following three types of results for each enrolled positive sample: Candidate test individual sample testing result (Candidateindividual), Candidate test pooled sample testing result (Candidatepool), and Comparator individual sample testing result (Comparatorindividual).
PPA between testing individual samples using the candidate test and testing individual samples using the comparator test (PPA, Candidateindividual vs. Comparatorindividual) should be estimated initially. If PPA (Candidateindividual vs. Comparatorindividual) is <95%, the candidate test should not be used to test pooled samples. If PPA (Candidateindividual vs. Comparatorindividual) is ≥95%, PPA between testing pooled samples using the candidate test and testing individual samples using the comparator test (PPA, Candidatepool vs. Comparatorindividual) should then be calculated as the percent of positive pools by the candidate test among 20 individual positive samples by the comparator test. When calculating PPA for the preliminary clinical sample pooling validation study, all non-negative results testing pooled samples should be counted as in agreement with positive individually tested results.
If PPA (Candidatepool vs. Comparatorindividual) is ≥80%, the n-sample pool is preliminarily validated. If PPA (Candidatepool vs. Comparatorindividual) is <80%, the test developer should repeat the preliminary validation study with smaller pool sizes to compensate for excessive loss in test sensitivity due to pooling at the larger sample size tested, until the PPA (Candidatepool vs. Comparatorindividual) for a smaller pool size is ≥80%. If there is sufficient volume, the same positive samples utilized in the evaluation of a larger pool size can be used in the validation of a smaller pool size. If ≥80% PPA (Candidatepool vs. Comparatorindividual) cannot be achieved for any pool sizes evaluated, the candidate test should not be used to test pooled samples.
To confirm that negative samples remain negative in n-sample pools, the test developer should also test in the preliminary clinical sample pooling validation study a sufficient number of individual negative samples, as determined by the candidate test, to generate at least 20 pools, each consisting of n negative samples for testing using the candidate test. For example, 100 negative samples are recommended to make up 20 5-sample negative pools (5x20 negatives). If there is sufficient volume, the same negative samples can be used to create positive and negative pooled samples.
Clinical Sample Pooling Validation Study with Samples from Three Geographically Diverse US Sites:
For tests intended to be performed at multiple laboratory sites (i.e., distributed tests), or at a single laboratory site receiving samples from geographically diverse sites, the below validation should be conducted to evaluate pooled testing in populations that may have different distributions of viral loads and different positivity rates.
Option A (appropriate for all tests):
The test developer should conduct an additional clinical sample pooling validation study with individual positive clinical samples at three geographically diverse sites in the US, assessing the PPA (Candidatepool vs. Candidateindividual) between testing n-sample pools (n is the preliminarily validated pool size) and assaying single samples using the candidate test. Each of the three sites should initiate n-sample pooling and enroll a minimum of 15 consecutive positive samples with sufficient volume for a minimum of 45 consecutive positive samples in total.
The validation study at each site should start from the starting time T0 and should consist of individual sample testing in parallel with the pooled testing. However, since all non-negative sample pools require testing of all individual samples included in the pool as a part of the n-sample pooling and deconvoluting workflow, the validation study essentially adds testing individual samples from the negative n-sample pools.
The validation study at each site may conclude at time T1, when a minimum of 15 consecutive positive individual results are obtained, including both positive individual results generated from individual testing of samples from the non-negative sample pools following the n-sample pooling and deconvoluting workflow, and positive individual results obtained from individual testing of samples from the negative sample pools for the time period from T0 to T1 [T0, T1].
Defining the number of positive individual sample results among negative sample pools as K, PPA between testing n-sample pools (n is the preliminarily validated pool size) and individual samples using the candidate test at each site should be calculated as PPA (Candidatepool vs. Candidateindividual) = 100% x (15-K)/15. It is critical that all consecutive positive samples from time period [T0, T1] are included in the PPA calculations. When calculating PPA for this sample pooling validation study, all non-negative results testing pooled samples should be counted as in agreement with positive individually tested results. Test developers should present PPA (Candidatepool vs. Candidateindividual) for each of three geographically diverse sites in the US separately.
Acceptance Criteria for Option A of the Clinical Sample Pooling Validation Study at Three Geographically Diverse US Sites:
If PPA (Candidatepool vs. Candidateindividual) for each site is ≥85%, the validation data supports n-sample pooling.
If PPA (Candidatepool vs. Candidateindividual) is ≥85% at 2 out of 3 sites, at the site where PPA (Candidatepool vs. Candidateindividual) is <85%, additional data with at least 15 additional consecutive positive samples should be generated and an estimate of the PPA (Candidatepool vs. Candidateindividual) for the combined data of at least 30 consecutive positive samples should be calculated with an overall PPA target of ≥85%.
If PPA (Candidatepool vs. Candidateindividual) is ≥85% at 0 or 1 out of 3 sites, or 1 site has PPA (Candidatepool vs. Candidateindividual) <85% calculated with a combined data of at least 30 consecutive individual positive samples, the validation data does not support n-sample pooling and a new decreased n (e.g., n-1) should be considered and validated.
Option B (appropriate for RT-PCR tests that can generate Ct values):
As an alternative and potentially less burdensome approach for assessing the PPA (Candidatepool vs. Candidateindividual) at three geographically diverse sites in the US using the candidate RT-PCR test that can generate Ct values, the test developer may acquire historical individual sample testing positive results from three geographically diverse sites in the US to perform in silico analysis of PPA (Candidatepool vs. Candidateindividual) for each site. We recommend acquiring at least 30 consecutive individually positive sample results by the candidate test (could be recently acquired historical data) from each of the three sites, for a minimum of 90 consecutive individually positive sample results in total.
In order to conduct in silico PPA (Candidatepool vs. Candidateindividual) analyses, for each candidate test target, the test developer should estimate the Ct shift that corresponds to n-sample pools (dilution of 1:n) using both the wet testing data generated from the preliminary clinical sample pooling validation study and additional wet testing of dilutions of a positive clinical sample in replicates using the candidate test at n x LoD, LoD and LoD/n, as described below:
Assess LoD by testing serial dilutions of a positive clinical sample in pooled negative clinical samples;
Test 5 replicates of this positive clinical sample at a concentration of n x LoD (designated as sample A);
Test 10 replicates of sample A combined with (n-1) individual negative clinical samples at the LoD concentration (designated as sample B);
Test 10 replicates of sample B combined with (n-1) individual negative clinical samples at the concentration of LoD/n (designated as sample C). Percent of positive results for sample C is designated as %D.
Recommendations regarding estimating the Ct shift corresponding to n-sample pools (dilution of 1:n) are provided below:
Construct and present a scatter plot of Ct pool (Y-axis) vs Ct individual (X-axis) using the data from wet testing the 20 positive clinical samples in the preliminary clinical sample pooling validation study, and the first replicate of sample A (Sample A Ct individual) and first replicate of sample B (Sample A Ct pool).
Divide the data points into 3 subintervals based on individual Ct values: ~7 points of high Ct values, ~7 points of medium Ct values, and ~7 points of low Ct values.
Calculate the difference between Ct pool (Y-axis) vs Ct individual (X-axis) for each data point and calculate the average of differences for each subinterval as Average Low Ct Values, Average Medium Ct Values, Average High Ct Values.
Analyze visually whether there is a tendency that the average of the differences for high individual Ct values is larger than the average of the differences for low individual Ct values.
If such tendency is observed, use Average High Ct Values as the estimate of Ct shift for the n-sample pools. If such tendency is not observed, take an average of the differences of all 21 samples, and use this average value as the estimate of Ct shift for the n-sample pools.
Using the estimated Ct shift for the n-sample pools (Ct Shift) and data from the wet testing of dilutions of a positive clinical sample in replicates at n x LoD, LoD, and LoD/n, the following rules for in silico PPA (Candidatepool vs. Candidateindividual) analyses for each candidate test target can be established:
Interval of individual Ct values |
Percent of Detected in n-Sample Pools |
[Cutoff Ct, Cutoff Ct – Ct Shift] |
0% detected |
[Cutoff Ct – Ct Shift, Ct at the LoD], |
(D+0)/2 % detected |
[Ct at the LoD, Ct at the LoD - Ct Shift] |
(95+D)/2 % detected |
[Ct less than Ct at the LoD - Ct Shift] |
100% detected |
Test developers should calculate and present PPA (Candidate pool vs. Candidate individual) in silico for each of the three geographically diverse sites in the US separately.
To lessen the burden of individual laboratory customers intending to utilize the candidate test for testing pooled samples after FDA authorization, with regard to continued monitoring after sample pooling implementation, we strongly recommend that test developers perform preliminary clinical sample pooling validations and wet testing of dilutions of a positive clinical sample in replicates at n x LoD, LoD, and LoD/n for all pool sizes that are less than or equal to n (i.e., n = 5, 4, 3, and 2) to characterize the reduction in test analytical sensitivity (i.e., shift in Ct values and the percent of individual positive samples with low viral load that may be missed due to sample pooling) with respect to each of the pool sizes. The goal of the validation is to generate the following reference table to be included in the instructions for use:
A reference table containing rules for in silico PPA (Candidatepool vs. Candidateindividual) analyses for each candidate test target for all validated pool sizes, so that each laboratory that intends to utilize the assay for testing pooled samples may utilize these rules to perform in silico PPA analyses as part of a continued monitoring plan after sample pooling implementation. See Appendix B for more information regarding the pooling re-assessment.
Acceptance Criteria for Option B of the Clinical Sample Pooling Validation Study at Three Geographically Diverse US Sites:
If the PPA (Candidatepool vs. Candidateindividual) in silico for each site is ≥85%, the historical data supports n-sample pooling.
If PPA (Candidatepool vs. Candidateindividual) in silico is ≥85% at 2 out of 3 sites, at the site where PPA (Candidatepool vs. Candidateindividual) in silico is <85%, additional recent historical individual testing data from at least 30 additional consecutive positive samples should be acquired and an estimate of the PPA (Candidatepool vs. Candidateindividual) in silico for the combined data of at least 60 consecutive positive samples should be calculated.
If PPA (Candidatepool vs. Candidateindividual) in silico is ≥85% at 0 or 1 out of 3 sites, or 1 site has PPA (Candidatepool vs. Candidateindividual) in silico <85% calculated with a combined data of at least 60 consecutive individual positive samples, the in silico validation data does not support n-sample pooling and a new decreased n (e.g., n-1) should be considered and validated.
Appendix B: Pooling Implementation and Monitoring
The recommendations in this appendix use media pooling as the basis for examples. However, the concepts also apply to swab pooling.
Pooling Implementation (Laboratory Monitoring Part A):
Prior to implementation of pooled testing using a test authorized for such indication, a laboratory should determine the appropriate pool size based on percent positivity rate in the testing population and pooling testing efficiency. Test developers should include directions in their instructions for use to enable laboratories to complete this step, such as using historical data and the information included in Table 2, as described below.
A.1 If Historical Data for Individual Samples are Available
Positivity Rate of Individual Testing
Estimate positivity rate (Pindividual) in the laboratory based on individual sample testing, considering the previous 7-10 days. Pindividual is the number of positive results divided by the total number of tested patients during these 7-10 days.
Selection of n for n-sample pooling
n should never be higher than the n validated by the test developer and included in the EUA-authorized instructions for use.
Use Pindividual and Table 2 to choose an appropriate validated pool size. Table 2 presents the maximum efficiency for the validated pool sizes corresponding to different positivity rates. If the positivity rate (Pindividual) is in Table 2, choose n from Table 2 which corresponds to the maximum efficiency (F).
If P individual in your laboratory is not listed in Table 2, you should calculate the efficiency for the maximum n which was validated, using formula F=1/ (1+ 1/n-(1-P)n). For example, if P individual in your laboratory is 1% and the maximum n which was validated is 5, the efficiency F=4.02 for n=5. An F of 4.02 generally means that an average of 4,020 individual samples can be tested using only 1,000 tests. You can use the same formula to calculate the efficiency for smaller sample sizes with a particular P individual as well.
If P individual is greater than 25%, then pooling patient samples is not efficient and should not be implemented.
A.2 If Historical Individual Data for Individual Samples is Unavailable:
If historical data from the previous 7-10 days is unavailable, the maximum pool size validated in the EUA and any smaller pool sizes can still be implemented, as the EUA-authorized test has been validated for the maximum pool size sample pooling. However, note that without P individual, the laboratory may choose a pooling size that does not maximize pooling efficiency.
Table 2: Efficiency of pooling based on the positivity of SARS-CoV-2 RNA in individual samples (as an example)
Pindividual, percent of positive subjects in the tested population |
nmaxefficiency (n corresponding to the maximal efficiency) |
F (Efficiency of n-sample pooling corresponding to nmaxefficiency (a maximum increase in the number of tested patients when Dorfman n- pooling strategy used))
|
1% |
11 |
5.11 |
2% |
8 |
3.65 |
3% |
6 |
3.00 |
4% |
6 |
2.60 |
5% |
5 |
2.35 |
6% |
5 |
2.15 |
7% |
4 |
1.99 |
8% |
4 |
1.87 |
9% |
4 |
1.77 |
10% |
4 |
1.68 |
11% |
4 |
1.61 |
12% |
4 |
1.54 |
13% |
3 |
1.48 |
14% |
3 |
1.43 |
15% |
3 |
1.39 |
16% |
3 |
1.35 |
17% |
3 |
1.31 |
18% |
3 |
1.28 |
19% |
3 |
1.25 |
20% |
3 |
1.22 |
21% |
3 |
1.19 |
22% |
3 |
1.16 |
23% |
3 |
1.14 |
24% |
3 |
1.12 |
25% |
3 |
1.10 |
Pooling Monitoring (Laboratory Monitoring Part B):
After implementation of pooled testing using a test authorized for such indication, a laboratory should perform ongoing monitoring of the positivity rate of pooled samples (i.e., Pindividual is the historical positivity rate and Ppool = the positivity rate from pooled samples from a rolling 7-10 day* period) to ensure pooled testing remains efficient. Test developers should include directions in their instructions for use to enable laboratories to complete this monitoring, as described below.
* It is recommended that Pindividual be calculated from the previous 7-10 days (i.e., prior to implementing pooled testing), while Ppool is calculated from data collected during a rolling 7-10 day time frame. However, when determining if 7-10 days is appropriate, take into consideration the laboratory testing volume and percent positivity, among other factors. Note that if the number of individual or pooled positive results collected during a given time frame is less than 10, Pindividual and Ppool may not be representative of the percent positivity in the testing population and the laboratory may want to consider extending the time period to increase the chance of capturing positives.
B.1 Historical Data for Individual Samples is Available:
If historical data for individual samples is available, compare P pool to P individual periodically, where Pindividual is the historical positivity rate. If P pool is less than 85% of P individual (P pool < 0.85 × P individual), it is recommended that:
The n-sample pooling should be re-assessed by conducting a re-assessment study as described in “Pooling Re-assessment (Laboratory Monitoring Part C)” below.
Alternatively, if the EUA-authorized test is a high sensitivity RT-PCR assay which uses a chemical lysis step followed by solid phase extraction of nucleic acid (e.g., silica bead extraction), and has established high sensitivity with an internationally recognized standard or FDA SARS-CoV-2 Reference Panel, 29 the size of pools may be increased taking into consideration Table 2, and the new n should not be more than the test developer validated as the maximum n in the EUA.
If P pool is greater than 25%, pooling of patient samples is not efficient and should be discontinued until the percent positivity rate decreases.
B.2 Historical Data for Individual Samples is Unavailable:
If historical data for individual samples is not available, it is recommended that n-sample pooling be assessed as follows:
After implementing a n-sample pooling strategy, first calculate the positivity rate (P pool-initial) based on n-sample pool size using the data from testing pooled samples from the first 7-10 days.
If P pool-initial is greater than 25%, pooling of patient samples is not efficient and should be discontinued until the percent positivity rate decreases.
If P pool-initial is less than or equal to 25%, pooling of patient samples can be continued.
Continue to monitor n-sample pooling strategy by calculating the positivity rate among patient samples during n-sample pooling (P pools-x) for ongoing 7-10 day periods based on n-sample pool testing. (P pool-x) should be updated daily using a moving average.
Compare P pool-initial to P pool-x periodically. If P pool-x is less than 90% of P pool-initial (P pool-x < 0.90 × P pool-initial), it is recommended that:
The n-sample pooling should be re-assessed by conducting a re-assessment study, as described in “Pooling Re-assessment (Laboratory Monitoring Part C)” below.
Alternatively, if the EUA-authorized test is a high sensitivity RT-PCR assay which uses a chemical lysis step followed by solid phase extraction of nucleic acid (e.g., silica bead extraction), and has established high sensitivity with an internationally recognized standard or FDA SARS-CoV-2 Reference Panel, 30 the size of pools may be increased taking into consideration Table 2, and the new n should not be more than the test developer validated as the maximum n in the EUA.
If P pool is greater than 25%, pooling of patient samples is not efficient and should be discontinued until the percent positivity rate decreases.
Pooling Re-assessment (Laboratory Monitoring Part C):
If, during monitoring of the positivity rate of pooled samples, a laboratory determines a drop in the positivity rate, as discussed above, the laboratory should conduct a re-assessment study. Test developers should include directions in their instructions for use to enable laboratories to complete this step, as described below.
Option 1: Re-assess pooling using individually tested samples:
Pause n-sample pooling and return to individual testing.
Patient samples should be tested individually until 10 consecutive positive samples have been collected. The total number of samples, tested individually, depends on the positivity rate.
Using these samples, 10 pools should be created and tested with 1 positive and (n-1) negative samples and the PPA between testing sample pools and individual samples should be calculated.
Alternatively, if the laboratory is using an EUA-authorized RT-PCR test that can generate Ct values, the laboratory may be able to assess PPA (PPApool vs. PPAindividual) in silico based on the individually tested sample results without performing any testing of pooled samples. In order to perform this in silico PPA assessment, the instructions for use of this EUA-authorized test must include a reference table containing information regarding the in silico PPA (PPApool vs. PPAindividual) analysis rules established by the test developer for each test target for all validated pool sizes. Refer to the “Clinical Sample Pooling Validation Study at Three Geographically Diverse US Sites” section in Appendix A for more information regarding this reference table.
Option 2: Re-assess pooling using individual testing and n-sample pooled testing:
The re-assessment study should start from starting time T0 and should consist of individual sample testing in parallel with the pooled testing. However, since all non-negative sample pools should include individual testing of all individual samples included in the pool as a part of the n-sample pooling and deconvoluting workflow, the re-assessment study essentially consists of testing individual samples from the negative n-sample pools.
The re-assessment study may stop and assess at time T1 when a minimum of 10 consecutive positive individual results are obtained, including both positive individual results generated from individual testing of samples from the non-negative sample pools following the n-sample pooling and deconvoluting workflow, and positive individual results obtained from individual testing of samples from the negative sample pools for the time period from T0 to T1 [T0, T1].
Defining the number of positive individual sample results among negative pools as K, PPA between testing n-sample pools and individual samples using the EUA-authorized test should be calculated as PPA (PPApool vs. PPAindividual) = 100% x (10-K)/10. It is critical that all consecutive positive samples from time period [T0, T1] are included in the PPA calculations. With regard to calculating the PPA, all non-negative results testing pooled samples should be counted as in agreement with positive individually tested results.
Re-assessment Acceptance Criteria for Option 1 and Option 2:
If the PPA (PPApool vs. PPAindividual) is ≥ 90% (9 out of 10 or 10 out of 10), then implementation of testing using n-sample pooling is appropriate.
If the PPA between pooled-testing results and individual-testing results is less than 90%:
If PPA ≤70% (7 out of 10), reduce the pool size (consider a new n as n-1).
If PPA is 80% (8 out of 10), collect an additional 10 consecutive individually positive samples. Then, calculate the PPA from the combined data of 20 samples, between pooled testing results and individual testing results. If the PPA is ≥ 85%, then implementation of testing using n-sample pooling is appropriate. Or, to compensate for lost sensitivity, reduce the pool size (consider a new n as n-1) and continue with the re-assessment testing until PPA of pooled compared to individual testing is ≥ 90%.
If PPA of at least 85% cannot be reached for any pool size evaluated in the re-assessment, cease pooling patient samples.
If n-sample pooling is acceptable based on re-assessment, re-establish P individual in your laboratory by estimating the positivity rate from individual testing in the population from which the 10 (or 20) consecutive individual positive samples were collected. If the total number of samples (N*) that needed to be tested to obtain the 10 (or 20) consecutive positive samples is stopped at the 10th (or 20th) positive sample, then the positivity rate of 10/N* (or 20/N*) is overestimated. The positivity rate should be corrected by the following corresponding multiplier:
Positivity rate for 10 samples is (10/N*) × (10/11).
Positivity rate for 20 samples is (20/N*) × (20/21).
This updated new positivity rate should be used as P individual in the future laboratory monitoring (see “Pooling Monitoring (Laboratory Monitoring Part B)” above.
This section applies only to
the requirements of the Paperwork Reduction Act of 1995 The
burden time for this collection of information is estimated to
average 34 to 45 hours per response, including the time to review
instructions, search existing data sources, gather and maintain the
data needed and complete and review the collection of information.
Send comments regarding this burden estimate or any other aspect of
this information collection, including suggestions for reducing this
burden to the address to: Department
of Health and Human Services An
agency may not conduct or sponsor, and a person is not required to Food
and Drug Administration
respond
to, a collection of information unless it displays a currently Office
of Operations
valid
OMB control number. Paperwork
Reduction Act (PRA) Staff DO
NOT SEND YOUR COMPLETED FORM TO THE PRA STAFF EMAIL ADDRESS
1 This template is part of the “Policy for Coronavirus Disease-2019 Tests During the Public Health Emergency (Revised),” available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/policy-coronavirus-disease-2019-tests-during-public-health-emergency-revised.
2 Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/policy-coronavirus-disease-2019-tests-during-public-health-emergency-revised.
3 All EUA templates can be found at https://www.fda.gov/medical-devices/coronavirus-disease-2019-covid-19-emergency-use-authorizations-medical-devices/vitro-diagnostics-euas#covid19ivdTemplates.
4 Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/emergency-use-authorization-medical-products-and-related-authorities.
5 Available at https://www.fda.gov/media/152768/download.
6 The CDC has granted a right of reference to the performance data contained in the CDC's EUA request for the CDC 2019-nCoV Real-Time RT-PCR Diagnostic Panel (CDC) (FDA submission number EUA200001) to any entity seeking authorization for a COVID-19 diagnostic device. The CDC has also granted a right of reference to the performance data contained in the CDC's EUA request for their Influenza SARS-CoV-2 (Flu SC2) Multiplex Assay (FDA submission number EUA201781) to any entity seeking authorization for a multi-analyte respiratory panel that includes SARS-CoV-2. CDC has published the primer and probe sequences for the Influenza SARS-CoV-2 Multiplex Assay on the CDC website.
7 Available at https://www.cdc.gov/csels/dls/sars-cov-2-livd-codes.html (last accessed on July 7, 2021). Note this website is not controlled by FDA.
8 Available at https://www.hhs.gov/coronavirus/testing/covid-19-diagnostic-data-reporting/index.html (last accessed on July 24, 2021). Note this website is not controlled by FDA.
9 Available at https://www.hhs.gov/sites/default/files/hhs-guidance-implementation.pdf (last accessed on July 24, 2021). Note this website is not controlled by FDA.
10 Available at https://www.hhs.gov/sites/default/files/non-lab-based-covid19-test-reporting.pdf (last accessed on July 24, 2021). Note this website is not controlled by FDA.
11 Available at https://www.fda.gov/medical-devices/postmarket-requirements-devices/quality-system-qs-regulationmedical-device-good-manufacturing-practices.
12 Further information regarding cybersecurity is available at https://www.fda.gov/medical-devices/digital-health/cybersecurity.
13 Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/guidance-content-premarket-submissions-software-contained-medical-devices.
14 Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/general-principles-software-validation.
15 Available at https://www.fda.gov/medical-devices/digital-health-center-excellence/device-software-functions-including-mobile-medical-applications.
16 Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/shelf-software-use-medical-devices#:~:text=Off%2Dthe%2Dshelf%20(OTS,to%20run%20device%2Dspecific%20functions.
17 GISAID is a global science initiative and primary source that provides open-access to genomic data of influenza viruses and the novel coronavirus responsible for COVID-19 (See https://www.gisaid.org/ (last accessed on July 26, 2021). Note this website is not controlled by FDA.)
18 https://www.fda.gov/regulatory-information/search-fda-guidance-documents/policy-evaluating-impact-viral-mutations-covid-19-tests
19 Last accessed on July 9, 2021. Note this website is not controlled by FDA.
20 Available at https://www.fda.gov/medical-devices/coronavirus-covid-19-and-medical-devices/sars-cov-2-reference-panel-comparative-data.
21 https://www.fda.gov/regulatory-information/search-fda-guidance-documents/guidance-informed-consent-vitro-diagnostic-device-studies-using-leftover-human-specimens-are-not.
22 Available at https://www.fda.gov/media/146695/download.
23 All templates can be accessed at https://www.fda.gov/medical-devices/coronavirus-disease-2019-covid-19-emergency-use-authorizations-medical-devices/vitro-diagnostics-euas#covid19ivdTemplates.
24 Available at https://www.fda.gov/about-fda/cdrh-transparency/clia-waiver-application-decision-summaries.
25 Last accessed on July 6, 2021. Note this website is not controlled by FDA.
26 A list of EUA-authorized tests and their accompanying fact sheets are available at https://www.fda.gov/medical-devices/coronavirus-disease-2019-covid-19-emergency-use-authorizations-medical-devices/vitro-diagnostics-euas.
27 Report Adverse events, including problems with test performance or results, to MedWatch by submitting the online FDA Form 3500 (https://www.accessdata.fda.gov/scripts/medwatch/index.cfm?action=reporting.home) or by calling 1-800-FDA-1088
29 Please see the following website for the most recent list of FDA authorized SARS-CoV-2 molecular tests: https://www.fda.gov/medical-devices/coronavirus-disease-2019-covid-19-emergency-use-authorizations-medical-devices/in-vitro-diagnostics-euas-molecular-diagnostic-tests-sars-cov-2.
Please see the following website for the results of the FDA SARS-CoV-2 reference panel testing: https://www.fda.gov/medical-devices/coronavirus-covid-19-and-medical-devices/sars-cov-2-reference-panel-comparative-data.
30 Please see the following website for the most recent list of FDA authorized SARS-CoV-2 molecular tests: https://www.fda.gov/medical-devices/coronavirus-disease-2019-covid-19-emergency-use-authorizations-medical-devices/in-vitro-diagnostics-euas-molecular-diagnostic-tests-sars-cov-2.
Please see the following website for the results of the FDA SARS-CoV-2 reference panel testing: https://www.fda.gov/medical-devices/coronavirus-covid-19-and-medical-devices/sars-cov-2-reference-panel-comparative-data.
| File Type | application/vnd.openxmlformats-officedocument.wordprocessingml.document |
| File Title | Rev 1 |
| Author | Takai, Erica |
| File Modified | 0000-00-00 |
| File Created | 2025-12-05 |
© 2026 OMB.report | Privacy Policy