Reporting-FDAAA, Section 520(m)(6)(A)(ii)- Assignment of Annual distribution Number ( ADN) for Devices Indicated For Pediatric Patients
Guidance: Humanitarian Device Exemption Regulation: Questions and Answers
Guidance - HDE-Questions and Answers
Reporting-FDAAA, Section 520(m)(6)(A)(ii)- Assignment of Annual distribution Number ( ADN) for Devices Indicated For Pediatric Patients
OMB: 0910-0661
Contains Nonbinding Recommendations
Guidance for HDE Holders, Institutional Review Boards (IRBs), Clinical Investigators, and FDA Staff
Humanitarian Device Exemption (HDE) Regulation: Questions and Answers
Document issued on: [insert release date of FR Notice]
This document supersedes: Humanitarian Device Exemptions (HDE) Regulation: Questions and Answers, issued July 18, 2006.
The draft of this document was issued on August 5, 2008
OMB control number: 0910-xxxx
Expiration Date: xx/xx/xxxx
See additional PRA statement in Section 67 of this guidance
For questions regarding this document, contact Stephen Rhodes, CDRH, at (301) 796-5640 or stephen.rhodes@fda.hhs.gov or the Office of Communication, Outreach and Development (CBER) at 1-800-835-4709 or 301-827-1800.

U.S. Department of Health and Human Services
Food and Drug Administration
Center for Devices and Radiological Health
Center for Biological Evaluation and Research

Preface
Public Comment
Written comments and suggestions may be submitted at any time for Agency consideration to the Division of Dockets Management, Food and Drug Administration, 5630 Fishers Lane, Room 1061, (HFA-305), Rockville, MD, 20852. Alternatively, electronic comments may be submitted to http://www.regulations.gov. When submitting comments, please refer to Docket No. FDA-2008-D-0434. Comments may not be acted upon by the Agency until the document is next revised or updated.
Additional Copies
Additional copies are available from the Center for Devices and Radiological Health (CDRH) through the Internet at: http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm089753.htm You may also send an e-mail request to dsmica@fda.hhs.gov to receive an electronic copy of the guidance document or send a fax request to 301-847-8149 to receive a hard copy. Please use the document number (1668) to identify the guidance document you are requesting.
Additional copies of this guidance document are also available from the Center for Biologics Evaluation and Research (CBER), Office of Communication, Outreach and Development (HFM-40), 1401 Rockville Pike, Suite 200N, Rockville, MD 20852-1448, or by calling 1-800-835-4709 or 301-827-1800, or from the Internet at http://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/default.htm.
Table of Contents
HUD Designations and HDE Applications 3
FDA’s Review of HDE Applications 3
The Role of Institutional Review Boards (IRBs) 3
Guidance for HDE Holders, Institutional Review Boards (IRBs), Clinical Investigators, and FDA Staff
Humanitarian Device Exemption (HDE) Regulation: Questions and Answers
This guidance represents the Food and Drug Administration's (FDA’s) current thinking on this topic. It does not create or confer any rights for or on any person and does not operate to bind FDA or the public. You can use an alternative approach if the approach satisfies the requirements of the applicable statutes and regulations. If you want to discuss an alternative approach, contact the FDA staff responsible for implementing this guidance. If you cannot identify the appropriate FDA staff, call the appropriate number listed on the title page of this guidance.
Introduction
This guidance document answers commonly asked questions about Humanitarian Use Devices (HUDs) and applications for Humanitarian Device Exemption (HDE) authorized by section 510(m)(2) of the Federal Food, Drug, and Cosmetic Act (the Act). This guidance document reflects the additional requirements set forth in the Pediatric Medical Device Safety and Improvement Act of 2007.
For the purposes of this guidance, “you” refers to the HDE holder, the Institutional Review Board (IRB), or the clinical investigator depending upon how the question is asked and “we” refers to FDA.
FDA's guidance documents, including this guidance, do not establish legally enforceable responsibilities. Instead, guidances describe the Agency's current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word should in Agency guidances means that something is suggested or recommended, but not required.
Definitions
1. What is a Humanitarian Use Device (HUD)?
As defined in 21 CFR 814.3(n), a HUD is a “medical device intended to benefit patients in the treatment or diagnosis of a disease or condition that affects or is manifested in fewer than 4,000 individuals in the United States per year.”
2. What is a Humanitarian Device Exemption (HDE)?
A Humanitarian Device Exemption (HDE)is an application that is similar to a premarket approval (PMA) application, but is exempt from the effectiveness requirements of sections 514 and 515 of the Food, Drug, and Cosmetic Act (the Act). FDA approval of an HDE authorizes an applicant to market a Humanitarian Use Device (HUD), subject to certain profit and use restrictions set forth in section 520(m) of the Act. Specifically, as described below, HUDs cannot be sold for profit, except in narrow circumstances, and they can only be used in a facility after an IRB has approved their use in that facility, except in certain emergencies.
3. Who is an HDE holder?
An HDE holder is a person who obtains the Humanitarian Device Exemption (HDE) from FDA.
4. What does it mean to “use” a HUD?
The term “use” in this document, when unmodified, refer to the use of a HUD according to its approved labeling and indication(s) to treat or diagnose patients. When a HUD is being used in a clinical investigation (i.e., collection of safety and effectiveness data), the terms “investigational use” or “clinical investigation” will be used. A HUD may be studied in a clinical investigation in accordance with its approved indication(s) for a different indication, subject to the requirements described below. For more information on "use" versus "investigational use"/"clinical investigation" of a HUD, see questions 40-42 and "Figure 1: Decision Tree for IRB Review of HUDs" at the end of this guidance.
HUD Designations and HDE Applications
5. What is required in a request for HUD designation?
In accordance with 21 CFR 814.102(a), the applicant’s request must include:
a statement indicating that the applicant is requesting a HUD designation for a rare disease or condition, or a valid subset of the disease or condition
the name and address of the applicant
a description of the rare disease or condition for which the device is to be used, the proposed indication or indications for use of the device, and the reasons why such therapy is needed
a description of the device and a discussion of the scientific rationale for the use of the device for the rare disease or condition
documentation, with appended authoritative references, to demonstrate that the device meets the definition of 21 CFR 814.3(n).
See 21 CFR 814.102(a) for additional information on each of the above items.
6. When does FDA determine whether a device is eligible for designation as a HUD?
After all supportive materials have been received along with the applicant’s request for HUD designation, we determine whether the device is for a rare disease or condition that affects, or is manifested in fewer than 4,000 individuals in the United States (US) per year. In the case of a device used for diagnostic purposes, we also determine at that time whether the documentation demonstrates that fewer than 4,000 individuals per year would be subjected to diagnosis by the device in the United States (21 CFR 814.102(a)(5)).
The applicant should submit the request for a HUD designation before submitting an application for an HDE.
7. Can a device qualify for HUD designation if the affected patient population is fewer than 4,000 per year but there may be multiple contacts with the device for a single patient?
Yes. FDA recognizes that, in some cases, the number of contacts with the device may exceed one per patient. A device that involves multiple patient contacts may still qualify for HUD designation as long as the total number of patients affected, or in which the disease or condition is manifested, is less than 4,000 per year in the US. In the case of a device used for diagnostic purposes, it may also still qualify for HUD designation despite there being multiple contacts with the device by a single patient; the documentation must demonstrate that fewer than 4,000 individuals per year would be subjected to diagnosis by the device in the United States (21 CFR 814.102(a)(5)). That is, devices used in 4,000 or more patients a year to diagnose a subpopulation of less than 4,000 patients with a disease or condition would not be eligible for HUD designation (21 CFR 814.102(b)(3)(ii)).
8. What is required in an HDE application?
The applicant must include a copy of or reference to FDA’s HUD designation letter with the HDE application (21 CFR 814.104(b)(1)). Other contents required in an HDE application are described in detail in 21 CFR 814.104. This information enables FDA to determine whether the device meets the statutory criteria for a HUD set forth in section 520(m)(2) of the Act.
The Pediatric Medical Device Safety and Improvement Act of 2007 (Public Law 110-85) requires additional information in all original HDE applications, if such information is readily available. Specifically, it requires: a description of any pediatric subpopulations that suffer from the disease or condition that the device is intended to treat, diagnose, or cure; and the number of affected pediatric patients. See section 515A(a)(2) of the Act.1
9. Can you submit an HDE application if another comparable device is available to treat or diagnose the disease or condition?
We will consider an HDE application for any of the following:
no comparable device is available to treat or diagnose the disease or condition; or
a comparable device is available under another approved HDE application; or
a comparable device is being studied under an approved Investigational Device Exemption (IDE) (21 CFR 814.104(b)(2)).
However, we cannot approve an HDE for a HUD device once a comparable device with the same indications for use is marketed through either the premarket approval (PMA) process or the premarket notification (510(k)) process. See section 520(m)(2)(B) of the Act.
10. What does FDA consider a “comparable device”?
A “comparable device” need not be identical to the device submitted under the HDE application. In determining whether a comparable device exists, FDA will consider:
the device's indications for use and technological characteristics
the patient population to be treated or diagnosed with the device
whether the device meets the needs of the identified patient population.
Contact Information
11. Where do I submit a request for a HUD designation?
Submit 2 copies of your request for a HUD designation in accordance with 21 CFR 814.102 to:
Office of Orphan Products Development (OOPD), HF-35
Food and Drug Administration
5600 Fishers Lane
Rockville, MD 20857
If you have questions about the HUD designation, FDA’s Office of Orphan Products Development is available at (301) 827-3666.
12. Where do I submit an HDE application?
Submit 6 copies2 of your HDE application in accordance with 21 CFR 814.20 and 814.104 to:
For Products Regulated by CDRH
U.S.
Food and Drug Administration
Center for Devices and
Radiological Heath
Document Mail Center – WO66-0609
10903
New Hampshire Avenue
Silver Spring, MD 20993-0002.
For Products Regulated by CBER
Document Control Center (HFM-99)
Center for Biologics Evaluation and Research
Food and Drug Administration
1401 Rockville Pike, Suite 200N
Rockville, MD 20852-1448
FDA’s Review of HDE Applications
13. How long does FDA have to review an original HDE application?
FDA has 75 days from the date of receipt to approve or deny an HDE application under 21 CFR 814.114. This period includes a 30-day filing period during which we determine whether the HDE application is sufficiently complete to permit substantive review. If we notify the applicant that the application is incomplete and request additional information, the 75-day time frame will reset upon receipt of the additional information by FDA. See section 520(m)(2) of the Act; 21 CFR 814.114.
14. What are the review time frames for HDE amendments, supplements, and reports?
The review timeframe for HDE amendments, supplements, and reports is 75 days, the same as for HDE original applications, except for a supplement submitted as a 30-day notice (21 CFR 814.39(f)).
15. Are HDE amendments, supplements, and reports subject to the same regulations as those for PMAs?
Yes. HDE amendments, supplements, and reports are generally subject to the same regulations as those for PMAs. See 21 CFR 814.106, 814.108, 814.110, and 814.126 for specific HDE requirements.
16. Are HDEs subject to user fees?
No. User fees for HDEs are waived under the Medical Device User Fee and Modernization Act of 2002, as reauthorized and amended by the Medical Device User Fee Amendments of 2007.
17. Does the Quality Systems Regulation (QSR) (21 CFR Part 820) apply to HUDs?
Yes, however, we primarily focus on those manufacturing practices the agency deems most relevant to the safety of the device.
18. Can I request an exemption from the QSR?
Yes. If you believe that you cannot comply with or should not be held to the QSR requirements, you may request an exemption. As described in 21 CFR 820.1(e), the procedures for petitioning for an exemption are set forth in 21 CFR 10.30. In evaluating such a request, we will give overriding consideration to the risks posed by the device, the potential risks that a manufacturing defect might pose, and the public health need for the device.
HDEs and Pediatric Patients
19. If an HDE was approved for use in pediatric patients prior to the enactment of the Pediatric Medical Device Safety and Improvement Act of 2007, is the HDE holder prohibited from profiting from the sale of the device?
Yes, only original HDE applications for devices indicated for use in pediatric patients or in a pediatric subpopulation that are approved on or after September 27, 2007, are assigned an annual distribution number (ADN) and may be sold for profit (subject to restrictions described below). For example, an HDE supplement does not warrant eligibility for profit if the HDE was previously approved for use in pediatric patients or in a pediatric subpopulation.
20. Are separate HDE applications required for a device indicated for pediatric and adult use?
No. Devices that are intended to treat both a pediatric population and an adult population may be included in a single HDE application, but the indications for use should specify use in pediatric patients, or pediatric subpopulation(s), as well as use in adults. In some cases, the safety and probable benefit profile for devices intended for use in a pediatric population, or in a pediatric subpopulation, may differ from its use in an adult population. Therefore, it is recommended that HDE applications for devices intended for use in pediatric populations and adult populations include data supporting the use in both pediatric and adult populations.
We note that the Act, as amended by the Pediatric Medical Device Safety and Improvement Act of 2007 (Public Law 110-85), requires us to establish the annual distribution number (ADN) by assessing projected use of the product in “individuals,” a term that includes both pediatric and adult patients. See section 520(m)(6)(A)(ii) of the Act. This provision authorizes HDE holders to receive profit from the sale of HUDs that are indicated for pediatric use only, or for use in both pediatric and adult patients, subject to the upper limit of the ADN. In this way, when a device is potentially applicable to both pediatric and adult populations, the statute provides an incentive for an applicant to include in its HDE submission to FDA information establishing that the device will not expose pediatric patients to an unreasonable or significant risk of illness or injury and that the probable benefit to health from the use of the device outweighs the risk of injury or illness from its use. Such analysis should address the risks compared to the benefits, taking into account the probable risks and benefits of currently available devices or alternative forms of treatment. Only when a submission meets this standard for approval will FDA approve the product for use in pediatric patients, and only then will the HDE holder be eligible to receive profit from the sale of the device.
21. What is the annual distribution number (ADN) and how is it determined?
The Pediatric Medical Device Safety and Improvement Act of 2007 (Public Law 110-85) allows HUDs intended for use in pediatric patients or in a pediatric subpopulation and approved on or after September 27, 2007, to be sold for profit as long as the number of devices distributed in any calendar year does not exceed the annual distribution number (ADN). The ADN is determined by the agency when the agency approves the HDE. It is determined by estimating the number of individuals (pediatric and adult patients) affected by the disease or condition and likely to use the device each year multiplied by the number of devices reasonably necessary to treat each individual. If the number calculated is less than 4,000, then this number is the ADN. If the number calculated is equal to or more than 4,000, then the ADN is capped at 3,999 because the ADN must be less than 4,000 devices. See section 520(m)(6)(A)(ii) of the Act.
The applicant should provide supporting data for both the number of individuals likely to use the device each year, and the number of devices reasonably necessary to treat each such individual. The same principles that govern requests for a HUD designation, specifically documentation with appended authoritative references, should apply to requests for an ADN designation. See question 5 for more information on such documentation.
As stated in section 520(m)(8) of the Act, the agency’s Pediatric Advisory Committee will annually review all HUDs intended for use in pediatric patients that are approved on or after September 27, 2007, to ensure that the HDE remains appropriate for the pediatric populations for which it is approved.
22. After an HDE is approved and an ADN has been assigned, can an HDE holder request to have the ADN modified?
Yes. An HDE holder may submit an HDE supplement (21 CFR 814.108) requesting modification of the ADN based on new information regarding the number of individuals affected by the disease or condition. Again, the ADN must be less than 4,000.
23. Do HDE holders with ADNs set by the agency have special reporting requirements?
HDE holders assigned an ADN must immediately notify the agency if the number of devices distributed in a year exceeds the ADN. See section 520(m)(6)(A)(iii) of the Act. FDA interprets this statutory requirement to mean that HDE holders must immediately notify the agency by submitting an HDE report whenever the number of devices shipped, or sold, in a year, however they are used, exceeds the ADN.3 In this way, the new statutory notification requirement is generally consistent with the reporting requirement in 21 CFR 814.126(b)(1)(iii) discussed in the “After FDA Approves an HDE” section below (question 32): both concern the number of devices shipped or sold, however the devices are ultimately used (even if outside their approved indications). The only difference is that the new statutory provision requires immediate notification when the number shipped or sold in a year exceeds the ADN, whereas the current regulations require periodic reports on a timeframe specified in the HDE approval order.
In those rare cases in which a device holds both an HDE approval for a certain indication, and a PMA approval for a different indication, sales or shipments of the device pursuant to the PMA are not subject to the ADN reporting requirement. The ADN relates only to those devices that are on the market through the HDE process for a disease or condition that occurs in pediatric patients or in a pediatric subpopulation. In that instance, the manufacturer is only required to notify FDA when sales or shipments tracked pursuant to the HDE exceed the ADN.
24. What happens when the number of devices shipped or sold in a year exceeds the ADN?
For HUDs labeled for use in pediatric patients or in a pediatric subpopulation and approved on or after September 27, 2007, FDA exempts a certain number of these devices each year -- known as the ADN -- from the prohibition on profit (see questions 29 and 30 for more on this prohibition). It is the HDE holder's responsibility to immediately notify the agency in the form of an HDE report (21 CFR 814.126) when the number of HUDs shipped or sold in a year, however they are used, exceeds the ADN. Once this notification occurs, or once FDA discovers through an inspection that the ADN has been exceeded, then the general prohibition on profit applies for the remainder of the year. See section 520(m)(6)(D) of the Act.
25. If a device is manufactured in various sizes depending on a patient’s anatomy, the number of devices distributed may be more than the number of devices used in any year. Which number, the number used or the number distributed, is the ADN?
As described above, the ADN is the number of devices shipped or sold in a year that the agency exempts from the prohibition on profit. Once the HDE holder notifies the agency, or once the agency discovers through an inspection, that the ADN has been exceeded, sales of the device for the remainder of the year are subject to the general prohibition on profit. If the HDE holder ships multiple sizes, these shipments may or may not count toward the annual ADN tally, depending on whether these additional sizes are used or are returned to the HDE holder. (See footnote 3.)
26. What is the definition of pediatric patients?
As defined in section 520(m)(6)(E) of the Act, pediatric patients are patients who are 21 years of age or younger at the time of the diagnosis or treatment. A pediatric subpopulation means one of the following populations: neonates, infants, children, or adolescents. FDA reviews pediatric devices through all of its premarket pathways, including premarket notification (510(k)), premarket approval (PMA), biological license application (BLA), and humanitarian device exemption (HDE). Additional information about the definition of pediatric patients and pediatric use can be found in: “Guidance for Industry and FDA Staff: Premarket Assessment of Pediatric Medical Devices.”4
After FDA Approves an HDE
27. Is the HDE holder responsible for ensuring that their HUD is administered only in facilities with IRBs?
Yes. As stated in 21 CFR 814.124(a), the HDE holder is responsible for ensuring that a HUD is administered only in facilities having properly constituted and functioning IRBs. If the local IRB defers the HDE review to another similarly constituted IRB (such as an independent IRB), the local IRB must submit this delegation in a letter to the HDE holder. This submission keeps the HDE holder informed of all reviewing IRBs.
28. Is the HDE holder required to submit to FDA the names and addresses of the IRBs that approved the use of a HUD?
No. The applicant is not required to submit the names and addresses of the reviewing IRBs to FDA. However, as required in 21 CFR 814.126(b)(2), the applicant must maintain records of:
the names and addresses of the facilities to which the HUD was shipped
correspondence with reviewing IRBs
any other information required by a reviewing IRB or FDA.
29. Does the general prohibition on profit apply to HUDs even when used outside their approved indications?
HUDs, even when used outside their approved indications, are subject to the general
prohibition on profit. See section 520(m)(3) of the Act; 21 CFR 814.104(b)(5).5 As explained in the “HDEs and Pediatric Patients” section above, however, some HUDs are exempt from this prohibition if they are indicated for use in pediatric patients, or in a pediatric subpopulation, or for use in both pediatric and adult patients, subject to the upper limit of the ADN.
For devices that have both an HDE and a PMA approval for a different indication, there is no restriction on profit from sales pursuant to the PMA.
30. How should the HDE holder verify that the amount charged for the device does not exceed the costs of research and development, fabrication, and distribution?
If the HDE holder charges more than $250 for the device, FDA requires a report by an independent certified public accountant (CPA), or an attestation by a responsible individual of the HDE holder’s organization, verifying that the amount does not exceed the costs of research, development, fabrication, and distribution (21 CFR 814.104(b)(5)). If the amount charged is $250 or less, this requirement is waived. HDEs for pediatric use approved on or after September 27, 2008, are exempt from the prohibition against profiting from the sale of the device up to ADN, as explained in the "HDEs and Pediatric Patients" section above.
31. What adverse event reporting requirements apply to HUDs?
Device user facilities and manufacturers are required to submit medical device reports to FDA and to the “IRB of record” (i.e., the IRB approving the use of the HUD) (See sections 519(a) and (b) of the Act; 21 CFR 803.30, 803.50, and 814.126(a)). Among these requirements, manufacturers must submit reports to FDA and the IRB of record whenever a HUD may have caused or contributed to a death or serious injury, or has malfunctioned and would be likely to cause or contribute to a death or serious injury if the malfunction were to recur (21 CFR 803.50 and 814.126(a)). User facilities must submit reports to FDA, the IRB of record, and the manufacturer whenever a HUD may have caused or contributed to a death, and must submit reports to the manufacturer (or to FDA and the IRB of record if the manufacturer is unknown) whenever a HUD may have caused or contributed to a serious injury (21 CFR 803.30 and 814.126(a)). Serious injury means an injury or illness that (1) is life-threatening, (2) results in permanent impairment of a body function or permanent damage to a body structure, or (3) necessitates medical or surgical intervention to preclude permanent impairment of a body function or permanent damage to a body structure (21 CFR 803.3). Note: Pediatric adverse events will be reviewed periodically by the agency’s Pediatric Advisory Committee (http://www.fda.gov/oc/advisory/default.htm). The specific requirements for this reporting are set forth in the Medical Device Reporting (MDR) Regulation, at 21 CFR Part 803
32. What does the HDE holder need to provide to FDA in its periodic report with respect to the HUD designation?
You must provide us with updated information on a periodic basis demonstrating that the HUD designation is still valid, based on the most current and authoritative information available (21 CFR 814.126(b)). As part of these reporting requirements, you must report the number of devices shipped or sold since initial HDE marketing approval (21 CFR 814.126(b)(1)(iii)). FDA interprets this regulation to require HDE holders to report the total number of devices shipped or sold, no matter how they are used (whether for the approved indication(s), emergency use, or otherwise). However, for devices that have both an HDE approval and a PMA approval for a different indication, you are only required to report on the number of devices that are shipped or sold pursuant to the HDE, unless specifically required by the PMA Approval Order. The required frequency for these periodic reports is specified in each HDE approval order, as explained in 63 Fed. Reg. 59217, 59218 (Nov. 3, 1998).
If, based on information contained in these reports, we believe that the HUD designation may no longer apply to your device, we may contact you for additional information. See 21 CFR 814.126(b)(1) for more information on these reports.
33. Can an HDE holder submit an HDE supplement for a new indication for use of an approved HUD?
No. If you are seeking a new indication for use of an approved HUD, you must first obtain a HUD designation for the new indication for use and then submit a new original HDE application. In the new application, any information or data submitted in the HDE for the original indication may be incorporated by reference. See 21 CFR 814.110.
34. What happens to an approved HDE if, subsequently, FDA makes the determination that the disease or condition affects or is manifested in 4,000 or more individuals in the US per year?
If we make the determination that 4,000 or more individuals in the US are affected or manifest a certain disease or condition per year, we may consider whether the HDE should be withdrawn. We intend to consider factors such as the number of patients with the disease or condition, the feasibility of conducting a pivotal clinical trial (to demonstrate reasonable assurance of safety and effectiveness), and the public health need for the device.
35. If a HUD is being investigated in an IDE study for a different indication, does it impact the number of allowable patients under the HDE?
No. Investigational use of a HUD in an IDE study for a different indication does not impact the HDE approval. The HUD is intended for use in the treatment or diagnosis of a disease or condition that affects or is manifested in fewer than 4,000 individuals in the United States per year. The device being investigated in the IDE study for possible subsequent PMA approval or 510(k) clearance will not be for the same indications for use as the HUD.
36. After FDA approves an HDE for a HUD, if FDA subsequently approves a PMA or clears a 510(k) for the device or another comparable device with the same indication, what is the status of the HDE approval?
If we subsequently approve a PMA or clear a 510(k) for the HUD or another comparable device with the same indication, we may withdraw the HDE. Once a comparable device becomes legally marketed through PMA approval or 510(k) clearance to treat or diagnose the disease or condition in question, there may no longer be a need for the HUD and so the HUD may no longer meet the requirements of section 520(m)(2)(B) of the Act.
The Role of Institutional Review Boards (IRBs)
37. What are the differences between an HDE and an IDE? They both use “device exemption” in their titles and can thus be confusing to IRBs.
Quite simply, the term “exemption” for the HDE means that certain statutes and regulations need not be followed in order to legally market a HUD. An HDE approval is based on safety and probable benefit; HDEs are exempt from the requirement to provide a reasonable assurance of effectiveness, as otherwise required in sections 514 and 515 of the Act.
The term “exemption” for the IDE means certain statutes and regulations need not be followed in order to study an unapproved or uncleared device (or an approved or cleared device for an unapproved or uncleared indication) in a research study involving humans (i.e., an IDE is an investigational exemption). With this exemption, the unapproved or uncleared device can be shipped and used in human research.
We remind IRBs that question 4 of this document makes a distinction between “use” of a HUD and “investigational use”/ “clinical investigation” of a HUD. The term “use” in this document, when unmodified, refers to the use of a HUD according to its approved labeling and indication(s). If a HUD is being used in a clinical investigation (i.e., collection of safety and effectiveness data), whether for its HDE-approved indication(s) or for a different indication, then this document refers to “investigational use” or “clinical investigation” of the HUD. Such investigational use is subject to the same requirements that apply to all FDA-regulated clinical studies, including 21 CFR Parts 50 (Protection of Human Subjects) and 56 (Institutional Review Boards). Additionally, if the HUD is being studied for a use other than its approved indication(s), the IDE regulations at 21 CFR Part 812 apply.. See questions 40-42.
For a schematic view of the difference between "use" and "investigational use"/"clinical investigation" of a HUD, please refer to “Figure 1: Decision Tree for IRB Review of HUDs” at the end of this guidance.
38. Should an IRB be concerned if there is a HUD approved for one indication, while the same device is being studied or marketed for another indication that does not qualify for an HDE?
No. As stated above, a HUD may be used in accordance with its approved indication(s) for use while the same device is being studied under an IDE for a different indication. Additionally, the same device can be approved or cleared for another indication without impacting the HDE.
39. What are the differences between a PMA, 510(k) and an HDE?
Three regulatory paths to the market for devices are via Premarket Approval (PMA), Premarket Notification (510(k)), and HDE.
A device with an approved PMA is approved for marketing based on valid scientific evidence and reasonable assurance that the device is safe and effective for its intended use. Once approved, it can be marketed and sold within its approved labeling. There are no restrictions on the price, and it can be used by anyone qualified to use the device.
A 510(k) device is cleared for marketing when the agency finds that it is at least as safe and effective, that is, substantially equivalent, to a legally marketed device that is not required to have a PMA. Using valid scientific evidence, submitters compare their device to one or more similar legally marketed devices, comparing the indications for use and technological characteristics. Once cleared, it can be marketed and sold in accordance with its labeling. There are no restrictions on the price, and it can be used by anyone qualified to use the device.
A device with an approved HDE is approved for marketing, but the approval is based on evidence of safety and probable benefit. The Act and implementing regulations exempt HUDs from the requirement to establish a reasonable assurance of effectiveness. The HUD is intended for use in the treatment or diagnosis of a disease or condition that affects or is manifested in fewer than 4,000 individuals in the US per year. The manufacturer of a HUD can make a profit, subject to the limit of the ADN, only if it is indicated for use in a pediatric population or subpopulation or for use in both pediatric and adult patients, was approved on or after September 27, 2007, and with certain other restrictions. (See the “HDEs and Pediatric Patients” section above for further discussion of this profit allowance.) Another important difference is that HUDs require IRB approval before being used at a facility. See sections 520(m)(3), (4), (6) of the Act; 21 CFR 814.124.
40. How does an IRB distinguish between the use of a HUD and the study of a HUD in a clinical investigation (i.e., research)?
Prior to the approval of an HDE application for a device, any studies conducted using the device must be under the IDE regulations (21 CFR Part 812). Once the HDE is approved, the following information applies if a clinical investigator or the HDE holder wants to conduct a clinical investigation using the HUD.
An HDE holder may collect safety and effectiveness data in a clinical investigation for the HDE-approved indication(s) without an IDE. As long as the HUD is being studied in accordance with the approved indication(s) described in labeling, the HUD, as such, is legally marketed and can be lawfully shipped without an IDE. See 21 CFR 812.1. IRB approval (21 CFR Part 56) and protection of human subjects (21 CFR Part 50) are still required for these studies because they are FDA-regulated clinical studies.
Clinical investigation of a HUD for a different indication must be conducted in compliance with the IDE regulations at 21 CFR Part 812, in addition to requiring IRB approval (21 CFR Part 56) and protection of human subjects (21 CFR Part 50). If the device is a significant risk device, an FDA-approved IDE is required. See 21 CFR 812.1, 812.20. To date, all HUDs have been significant risk devices requiring FDA-approved IDEs. See question 42 for more discussion of significant risk devices.
In short, IRB approval, informed consent, and additional safeguards for children (if applicable) are required for the clinical investigation (investigational use) of a HUD, whether the HUD is being studied for its HDE-approved indication(s) or for a different indication. These requirements are separate and distinct from the requirements that apply to the use of a HUD at a facility: as described in questions 43 and 59, IRB approval is required before a HUD is used at a facility to treat or diagnose patients and the IRB may require informed consent as part of such approval. In other words, just because an IRB has approved use of a HUD at a facility to treat or diagnose patients does not mean that the IRB has approved investigational use of the HUD (i.e., in a clinical investigation), for the collection of safety and effectiveness data. For more information on the difference between "use" of a HUD and "investigational use"/"clinical investigation" of a HUD, see “Figure 1: Decision Tree for IRB Review of HUDs” at the end of this guidance.
41. What if the HDE holder decides to collect safety and effectiveness data in a study to support a PMA for the HDE-approved indications?
As stated above, you may collect safety and effectiveness data to support a PMA for the HDE-approved indication(s) without an IDE. While the work done to collect such safety and effectiveness data to support a PMA constitutes a clinical investigation, FDA considers the study exempt from the requirement for an IDE as long as the HUD is used in accordance with its approved indication(s). IRB approval (21 CFR Part 56) and protection of human subjects (21 CFR Part 50) are still needed, however, as required for all FDA-regulated clinical studies. As noted above, the IRB approval, informed consent, and additional safeguards for children (if applicable) required for the clinical investigation/investigational use of a HUD are separate and distinct from the IRB approval and any consent associated with the use of the HUD. That an IRB has approved use of a HUD at a facility to treat or diagnose patients does not mean the IRB has approved investigational use of the HUD (i.e., in a clinical investigation), for the collection of safety and effectiveness data.
If you want to collect safety and effectiveness data for a use other than the HDE-approved indication(s), you must comply with the IDE regulations at 21 CFR Part 812 in addition to complying with the requirements for IRB approval (21 CFR Part 56) and protection of human subjects (21 CFR Part 50).
42. Does an IRB have to make the determination of a significant risk (SR) or non-significant risk (NSR) device (21 CFR 812.66) when it reviews a HUD?
When an IRB is deciding whether to approve use of a HUD at a facility (see questions 43-52), its review does not include an SR/NSR determination. As noted above, use of a HUD at a facility to treat or diagnose patients is not a "clinical investigation"; the HUD as such is legally marketed for use within its HDE-approved indication(s).
If an IRB receives a request to review a clinical investigation of a HUD (i.e., collection of safety and effectiveness data), and that clinical investigation concerns the HDE-approved indication(s), then again the IRB does not have make an SR/NSR determination in its review. FDA considers such investigations exempt from the IDE requirements in 21 CFR Part 812, as noted above. Nonetheless, the IRB still has to approve the clinical investigation under 21 CFR Part 56 and informed consent and additional safeguards for children (if applicable) are required under 21 CFR Part 50, as for all FDA-regulated clinical studies.
In contrast, if the IRB receives a request to review an application for an investigational study of the HDE for a different indication, then the IRB should be alert that this type of clinical investigation is subject to the IDE regulations at 21 CFR Part 812. To date, all HUDs when studied for uses other than their approved indication(s) have been SR devices requiring an FDA-approved IDE. See 21 CFR 812.20(a). In practice, most sponsors have obtained an IDE from FDA before beginning such studies, and so IRBs have not needed to make the SR/NSR determination (i.e., the sponsors already knew their device was an SR device). However, in the event that a sponsor seeks IRB approval for research of a HUD for a indication other than its approved indication(s) without first obtaining an FDA-approved IDE, then the IRB should make the SR/NSR determination as described in 21 CFR 812.66.
43. Is IRB approval required before the use of a HUD at a facility?
Yes. As stated in section 520(m)(4) of the Act, IRB approval is required before a HUD is used at a facility, with the exception of emergency use (see question 65). The IRB must have among its members (or consultants) the appropriate experience and expertise to perform a complete and adequate review of the use of a HUD at that institution (21 CFR 56.107(a)). In addition, a local IRB may defer in writing to another similarly constituted IRB that has agreed to assume responsibility for review of the use of the HUD. This deferral letter must be sent to the HDE holder, because the HDE holder is responsible for ensuring that a HUD is administered only in facilities in which the reviewing IRB is constituted and acting in accordance with 21 CFR Part 56 (21 CFR 814.124(a)). See question 46 for further discussion of the scope of IRB approval.
44. Who is responsible for submitting materials to and obtaining approval from the IRB before the HUD is used at a facility?
As explained above, the HDE holder is responsible for ensuring that the HUD is administered only in facilities with properly constituted and functioning IRBs (see question 27). The health care provider at such facilities should be responsible for obtaining IRB approval before use of the HUD, except in certain emergencies where prior IRB approval is not required (see question 65). The IRB should have policies and procedures in place for receipt and evaluation of the materials necessary for initial approval and continuing review of the HUD.
45. How should an IRB evaluate requests for approval of the use of a HUD?
As stated in 21 CFR 814.124(a), an IRB that reviews and approves the use of a HUD must be constituted and act in accordance with the agency’s regulation governing IRBs (21 CFR Part 56), which include initial and continuing review of the use of the device. FDA recommends that an IRB follow the review criteria at 21 CFR 56.111 and elsewhere in Part 56 as much as possible. For example, you should review the risks to patients that are found in the product labeling, ensure the risks are minimized, and evaluate whether the risks are reasonable in relation to the proposed use of the device.
Specifically, FDA recommends reviewing the following materials during initial review of the HUD: a copy of the HDE approval order; a description of the device; the product labeling; the patient information packet that may accompany the HUD; a sample consent form for the use of the HUD, if required by the IRB; and a summary of how the physician proposes to use the device, including a description of any screening procedures, the HUD procedure, and any patient follow-up visits, tests or procedures. A list of approved HDEs may be found at http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfHDE/HDEInformation.cfm#2.
The approval order, labeling, and patient information may be found by selecting the number of the appropriate HDE. You should have policies and procedures in place for this review and approval, including whether your IRB requires a consent document for the use of the HUD.
46. To what extent should an IRB exercise oversight of clinician responsibilities in the use of a HUD?
In reviewing the use of the HUD, IRBs should be cognizant that the FDA has made a determination of safety and probable benefit for use of the HUD only within its approved indication(s). The IRB is not required to review and approve each individual use of a HUD. Rather, the IRB may use its discretion to determine how to approve use of a HUD. For example, if it so wishes, with the input of members with the appropriate expertise in the clinical area (21 CFR Part 56), an IRB may specify limitations on the use of the device based upon one or more measures of disease progression, prior use and failure of any alternative treatment modalities, reporting requirements to the IRB or IRB chairperson, appropriate follow-up precautions and evaluations, or any other criteria it determines to be appropriate.
47. What types of review functions are IRBs responsible for with respect to HUDs?
IRBs are responsible for initial as well as continuing review of the HUD. For initial review of a HUD, IRBs are required to perform their review at a convened meeting (21 CFR 56.108). For continuing review, IRBs may use the expedited review procedures (21 CFR 56.110). When applicable, review of the use of a HUD and review of the investigational use of a HUD in a clinical investigation may be done simultaneously.
48. Why does FDA suggest that an IRB perform the continuing review of a HUD using an expedited procedure?
FDA recommends the use of an expedited procedure because a HUD is a legally marketed device and no safety and effectiveness information is being collected systematically, as is required for a research protocol. An expedited review does not mean a less than substantive review. During the expedited review, the Chair or the Chair’s designated member(s) should thoughtfully consider the risk and benefit information available and any Medical Device Reporting (MDR) reports (see question 50). IRBs may develop their own policies and procedures for continuing review of a HUD and may perform this review at a convened meeting.
49. Should other committees at an institution be involved in the review of a HUD?
There is no regulatory requirement for committees other than the IRB to approve the use of a HUD. However, the institution may require additional review. For example, the use of another committee to provide assessments of specific risk posed by the technology or software compatibility may supplement the IRB review.
50. What does an IRB have to know about Medical Device Reporting (MDR)?
The HDE regulation, 21 CFR 814.126(a), requires that MDR reports submitted to FDA, in accordance with 21 CFR Part 803 (see question 31) shall also be submitted to the "IRB of record" (i.e., the IRB approving the use of the HUD).
51. What should an IRB consider with respect to the health care provider(s) who will use the HUD?
The IRB may want to ensure that health care providers are qualified through training and expertise to use the device. For many HDEs, the HDE holder is required to provide training on the use of the device prior to the health care provider using the device. Such requirements would be specified in the HDE approval order, available at http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfHDE/HDEInformation.cfm#2 (select the HDE number).
52. Must an IRB request a protocol to review for the use of a HUD?
When a HUD is used to treat or diagnose patients, i.e., not for research, we do not require submission of a protocol to the IRB for review. However, your IRB or institution may require one under its own policies and procedures.
53. Does FDA require an IRB to monitor the number of uses per year of a HUD?
No. It is the responsibility of the HDE holder to monitor how many devices are distributed each year, and if that number exceeds 4,000, to provide an explanation and estimate of how the device is being used by patients. See 21 CFR 814.126(b)(1)(iii).
54. Must an IRB review or audit the medical record of patients who received a HUD?
No, we do not require you to audit medical records of patients who receive a HUD.
55. Should an IRB ask for justification of the charges for the HUD?
No. There is no requirement for the IRB to request a justification of the charges for the HUD. FDA reviews the financial information in the HDE holder’s initial application, and periodically thereafter.
56. Should an IRB be concerned if an HDE holder charges for a HUD?
HDE holders generally charge for the HUD that is used to treat or diagnose a patient. However, HUDs cannot be sold for a price that exceeds the costs of research and development, fabrication, and distribution of the device. The exception is if they are indicated for use in a pediatric population, or pediatric subpopulation, or for use in both pediatric and adult patients, were approved on or after September 27, 2007, and annual sales have not yet exceeded the ADN (as discussed in “HDEs and Pediatric Patients” section above). See sections 520(m)(4), (6) of the Act.
If a HUD is studied in a clinical investigation of a new use, the sponsor of the clinical investigation may not charge subjects or investigators a price larger than necessary to recover the costs of manufacture, research, development, and handling (21 CFR 812.7(b)). Any costs for which a subject in a clinical investigation is responsible should be clearly explained in the informed consent document (21 CFR 50.25(b)(3)).
57. Does an IRB function as a Data Monitoring Committee for a HUD?
No. You may, however, ask the HDE holder for copies of the safety information submitted to FDA in the periodic reports required by 21 CFR 814.126(b)(1). In this way, information that could have a bearing on human safety would be considered at the time of continuing review.
58. Do the requirements for review of a HUD change if an IRB has a Federal Wide Assurance (FWA) with the Office of Human Research Protections?
No. The use of a HUD is not research; rather, it is use of a legally marketed device. We describe your responsibilities in section 520(m) of the Act and in the implementing regulations at 21 CFR 814.124. We also offer guidance to you in this document. If, however, a HUD is used in a clinical investigation (see question 41), IRBs should follow their FWA requirements and their written procedures for FDA-regulated research.
59. What information should be given to patients before they receive a HUD, and should patients consent to the HUD use?
Neither the Act nor the regulations require informed consent from patients for the use of a HUD. An IRB may, however, choose to require informed consent that is consistent with the approved labeling when the IRB approves use of the HUD in a facility.
Most HDE holders develop patient information packets that generally contain a discussion of the potential risks and benefits of the HUD and any procedures associated with its use. If patient information packets are available, the IRB should ensure that physicians distribute them to patients prior to their receiving the HUD. Even when an institution requires patients to sign a written consent document that describes the use of the HUD (and which may provide similar information found in the HDE holder’s packet), the patient should always receive the HDE holder’s patient information packet. For HUD patient information packets, go to http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfHDE/HDEInformation.cfm#2 and select the HDE number. In addition to the above information, many institutions also require informed consent for the surgery or procedure related to the use of the HUD.
If a HUD is studied in a clinical investigation, the informed consent of the subject must be obtained in accordance with FDA regulations at 21 CFR Part 50 (see question 41).
60. If an IRB requires a written consent document for the use of a HUD, what information should be included?
It would be reasonable for the document to include much of the information found in the HDE holder’s patient information packet. If no patient information packet is available, you may consider including the following: an explanation that the HUD is designed to diagnose or treat the disease or condition described in the HDE labeling and that no comparable device is available to treat the disease or condition; a description of any ancillary procedures associated with the use of the HUD; a description of the use of the HUD; all known risks or discomforts; and an explanation of the postulated mechanism of action of the HUD in relation to the disease or condition. You should also include information reflecting the HUD status of the device, such as a sentence indicating that the effectiveness of this device for this use has not been demonstrated. The IRB may decide to include other information.
If the HUD is studied in a clinical investigation, the elements included in the informed consent document must conform to the requirements found in 21 CFR 50.25.
61. Is it appropriate for the HUD labeling and materials to include the phrase “FDA approved”? What other information must the labeling contain?
HUD labeling and materials must be truthful and not misleading. See section 502(a) of the Act. The labeling may state that the device is approved as a HUD for its intended use, but the labeling must also include the following statement clarifying that effectiveness has not been demonstrated: “Humanitarian Device. Authorized by Federal law for use in the [treatment or diagnosis] of [specify disease or condition]. The effectiveness of this device for this use has not been demonstrated.” See 21 CFR 814.104(b)(4)(ii) for more information on HUD labeling requirements.
62. What should IRBs tell physicians who want to study a HUD for a new indication?
Physicians who want to study a HUD for a new indication must submit an IDE application to FDA if the device is a significant risk device (see question 42). Physicians may be either the sponsor or investigator of the study or they may want to involve the HDE holder as the sponsor. The investigational use of a HUD under these circumstances is a clinical investigation and must be conducted in accordance with 21 CFR Parts 812, 50, 54, and 56.
63. Does the use of a HUD constitute treatment or research under the Health Insurance Portability and Accountability Act of 1996 (HIPAA)? Does the IRB need to waive a HIPAA authorization for the use or disclosure of protected health information related to the use of a HUD?
The Privacy Rule promulgated at 45 CFR Parts 160 and 164, Subparts A and E pursuant to HIPAA governs the use and disclosure of certain individually identifiable health information (protected health information). An entity that is covered by HIPAA (a covered entity) may use and disclose protected health information without the patient’s authorization if the use or disclosure is for the purpose of treatment. If the use or disclosure of protected health information is for the purpose of research, then the covered entity generally must obtain the patient’s authorization, unless an IRB or Privacy Board has determined that such an authorization is not necessary because the research satisfies certain waiver criteria.
The use of a HUD according to its approved labeling and indication is generally for treatment or diagnosis, even though such use requires IRB approval. If a HUD is being used according to its approved labeling and indication, and not in a clinical investigation, then protected health information about a patient may be used or disclosed for treatment or diagnostic purposes without the patient’s authorization under HIPAA.
If a HUD is being used in a clinical investigation, whether or not the use of the HUD is the subject of the investigation, then protected health information about a patient that is used or disclosed for purposes of the clinical investigation requires the patient’s authorization under the HIPAA Privacy Rule. The IRB may waive this authorization if certain waiver criteria are met.
64. Does reporting of safety and effectiveness data to the sponsor require a HIPAA authorization or does this activity fall under an FDA-related activity under 45 CFR 164.512(b) (public health reporting)?
Reporting HUD safety information to the sponsor does not require a HIPAA authorization since it falls under the permissive disclosure for FDA-related activities at 45 CFR 164.512(b)(iii).
Using HUDs in Emergency Use Situations
65. When can a HUD be used without prior IRB approval?
If a physician in an emergency situation determines that IRB approval for the use of the HUD at the facility cannot be obtained in time to prevent serious harm or death to a patient, a HUD may be used without prior IRB approval. The physician must report the emergency use within five days; provide written notification of the use to the IRB chair person including identification of the patient involved, the date of the use, and the reason for the use. See section 520(m)(4) of the Act; 21 CFR 814.124.
66. After an IRB approves the use of the HUD at the facility, can a physician use a HUD outside its approved indication(s) in an emergency or if the physician determines there is no alternative device for the patient's condition?
Physicians should be cognizant that FDA has made a determination of safety and probable benefit for use of the HUD only within its approved indication(s). If a physician wants to use a HUD outside its approved indication(s), FDA recommends that the physician obtain informed consent from the patient and ensure that reasonable patient protection measures are followed, such as devising schedules to monitor the patient, taking into consideration the patient's specific needs and the limited information available about the risks and benefits of the device. FDA further recommends that the physician submit a follow-up report on the patient’s condition to the HDE holder and first check with the IRB before such use to review any institutional policy. The extent of IRB oversight in these circumstances is up to the IRB (see question 46). Note: as discussed in question 31, MDR reports must be submitted to FDA and to the “IRB of record” (i.e., the IRB approving the use of the HUD) if the device may have caused or contributed to death or serious injury and for certain malfunctions.
Figure 1: Decision Tree for IRB Review of HUDs
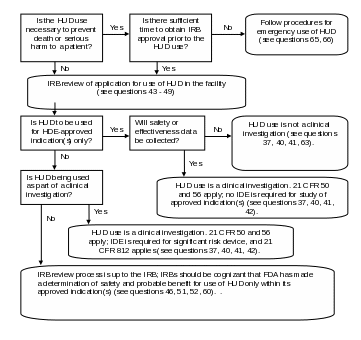
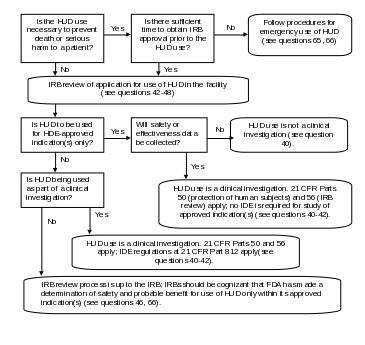
Note: Medical device reporting is required under 21 CFR Part 803 whenever the use of a HUD may have caused or contributed to a death or serious injury, or has malfunctioned and would be likely to cause or contribute to a death or serious injury if the malfunction were to recur (see questions 31, 50, 66). For investigational use of a HUD under an IDE, reports of unanticipated adverse device effects must be reported under 21 CFR 812.150(a)(1) and 812.150(b)(1).
Section 508 text for Figure 1.
Flowchart. Is the HUD use necessary to prevent death or serious harm to a patient? If no, proceed to node 1; if yes, is there sufficient time to obtain IRB approval prior to the HUD use? If yes, proceed to node 1; if no, Follow procedures for emergency use of HUD (see questions 65, 66). Node 1, IRB review of application for use of HUD in the facility (see questions 42-48). Is HUD to be used for HDE-approved indication(s) only? If no, proceed to node 2; if yes, will safety or effectiveness data be collected? If yes, HUD use is a clinical investigation. 21 CFR Parts 50 (protection of human subjects) and 56 (IRB review) apply; no IDE is required for study of approved indication(s) (see questions 40-42). If no, HUD use is not a clinical investigation (see question 40). Node 2, is HUD being used as part of a clinical investigation? If yes, HUD use is a clinical investigation. 21 CFR Parts 50 and 56 apply; IDE regulations at 21 CFR Part 812 apply (see questions 40-42). If no, IRB review process is up to the IRB; IRBs should be cognizant that FDA has made a determination of safety and probable benefit for use of HUD only within its approved indication(s) (see questions 46, 66).
Paperwork Reduction Act of 1995
This guidance contains information collection provisions that are subject to review by the Office of Management and Budget (OMB) under the Paperwork Reduction Act of 1995 (44 U.S.C. 3501-3520). The time required to complete this information collection is estimated to average 100 hours per response, including the time to review instructions, search existing data sources, gather the data needed, and complete and review the information collection. Send comments regarding this burden estimate or suggestions for reducing this burden to:
U.S. Food and Drug Administration
Center for Devices and Radiological Health
HDE Program WO66-1645
10903 New Hampshire Avenue
Silver Spring, MD 20993-0002
This draft guidance also refers to previously approved collections of information found in FDA regulations. The collections of information in 21 CFR part 803 have been approved under OMB control number 0910-0437; the collections of information in 21 CFR part 812 have been approved under OMB control number 0910-0078; the collections of information in 21 CFR part 807, subpart E have been approved under OMB control number 0910-0120; the collections of information in 21 CFR part
814, subparts A, B, and C have been approved under OMB control number 0910-0231; the collections of information in 21 CFR parts 50 and 56 have been approved under OMB control number 0910-0130; the collections of information in 21 CFR part 820 have been approved under OMB control number 0910-0073; the collections of information in 21 CFR part 814, subpart H have been approved under OMB control number 0910-0332; and the collections of information in 21 CFR 10.30 have been approved under OMB control number 0910-0183.
An agency may not conduct or
sponsor, and a person is not required to respond to, a collection
of information unless it displays a currently valid OMB control
number. The OMB control number for this information collection is
0910-xxxx, which expires on xx/xx/xxxx.
1 Many of the statutory provisions cited throughout this guidance, including sections 515A(a)(2) and 520(m)(6) of the Act, were added by the Pediatric Medical Device Safety and Improvement Act of 2007.
2 We encourage submission of electronic copies. For more information on submission of electronic copies to CDRH, please see "Electronic Copies for Pre-Market Submissions," http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/HowtoMarketYourDevice/PremarketSubmissions/ucm134508.htm. For electronic copies submitted to CBER, please see “Regulatory Submissions in Electronic Format for Biologic Products,” http://www.fda.gov/BiologicsBloodVaccines/DevelopmentApprovalProcess/ucm163685.htm.
3 FDA recognizes that HDE holders may ship additional sizes to facilities to ensure that each device fits properly when used. These additional shipments may or may not count towards the annual ADN tally, depending on whether these additional sizes are used or are returned to the HDE holder.
4 http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM089742.pdf
5 As discussed in a preamble to the HDE Regulation, "an applicant will not be considered in violation of [section 520(m)(3) of the Act] if [the applicant] receives incidental profits which exceed its good faith estimate of costs." 61 Fed. Reg. 33232, 33242 (June 26, 1996) (citing legislative history).
| File Type | application/msword |
| File Title | Draft Guidance for HDE Holders, Institutional Review Boards (IRBs), Clinical Investigators, and FDA Staff |
| Author | pchao |
| Last Modified By | DPresley |
| File Modified | 2010-02-24 |
| File Created | 2010-02-24 |
© 2026 OMB.report | Privacy Policy