eCopy Guidance
eCopy Guidance.doc
Guidance: Humanitarian Device Exemption Regulation: Questions and Answers
eCopy Guidance
OMB: 0910-0661
Contains Nonbinding Recommendations
Draft – Not for Implementation
eCopy Program for Medical Device Submissions
Guidance for Industry and Food
and Drug Administration Staff
Document issued on:
The draft of this document was issued on October 17, 2012.
For questions regarding this document, contact CDRH’s Office of Device Evaluation at 301-796-6055 or CBER’s Office of Communication, Outreach and Development at 1-800-835-4709 or 301-827-1800.


U.S. Department of Health and Human Services
Food and Drug Administration
Center for Devices and Radiological Health
Center for Biologics Evaluation and Research
Preface
Public Comment
You may submit written comments and suggestions at any time for Agency consideration to the Division of Dockets Management, Food and Drug Administration, 5630 Fishers Lane,
rm. 1061, (HFA-305), Rockville, MD, 20852. Submit electronic comments to http://www.regulations.gov. Identify all comments with the docket number listed in the notice of availability that publishes in the Federal Register. Comments may not be acted upon by the Agency until the document is next revised or updated.
Additional Copies
Additional copies are available from the Internet. You may also send an e-mail request to dsmica@fda.hhs.gov to receive an electronic copy of the guidance or send a fax request to 301-827-8149 to receive a hard copy. Please use the document number (1797) to identify the guidance you are requesting.
Additional copies of this guidance document are also available from the Center for Biologics Evaluation and Research (CBER), Office of Communication, Training and Manufacturers Assistance (HFM-40), 1401 Rockville Pike, Suite 200N, Rockville, MD 20852-1448, or by calling 1-800-835-4709 or 301-827-1800, or from the Internet at http://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/default.htm.
Table of Contents
3. Are differences between the contents of an eCopy and paper submission acceptable? 3
4. For what submission types is an eCopy required? 3
6. How many copies of a submission are needed? 5
7. What are the processing steps for an eCopy? 7
a. What are the standards for an eCopy? 7
b. How do you know beforehand if an eCopy meets the standards? 7
c. What if there is another processing party involved? 8
d. What if this is a Third Party 510(k)? 8
e. What if this is a bundled submission? 8
f. How do you submit an eCopy to FDA? 9
g. How does FDA process an eCopy? 9
h. What do you provide to FDA if your submission was placed on eCopy hold? 9
i. How does FDA process a replacement eCopy? 10
j. When does review of a submission begin? 10
8. What if your device is regulated by CBER? 10
a. Will the new eCopy requirements apply? 10
b. Can you submit an electronic submission instead? 11
c. How do you prepare and submit an electronic submission to CBER? 11
Attachment 1 –Standards for eCopies 13
A. Cover Letter Requirements 13
B. Volume or Non-Volume Structure Requirements 13
C. Adobe Acrobat PDF File Requirements 16
D. Requirements for How to Add Non-PDF Files via “STATISTICAL DATA” and “MISC FILES” Folders 18
E. Bookmarks and Hyperlinks within PDFs 21
eCopy Program for Medical Device Submissions
Guidance for Industry and Food and Drug Administration Staff
The purpose of this guidance is to explain the new electronic copy (eCopy) Program for medical device submissions. Section 745A(b) of the Federal Food, Drug, and Cosmetic Act (FD&C Act), added by section 1136 of the Food and Drug Administration Safety and Innovation Act (FDASIA) (Pub. L. 112-144), requires the submission of eCopies with the issuance of this final guidance. This guidance describes how the Food and Drug Administration (FDA) is implementing the eCopy Program under section 745A(b) of the FD&C Act. The inclusion of an eCopy is expected to improve the efficiency of the review process by allowing for the immediate availability of an electronic version for review rather than relying solely on the paper version.
This guidance provides, among other things, the standards for a valid eCopy under section 745A(b)(2)(A) of the FD&C Act. In accordance with section 745A(b), submission types identified in this final guidance must include an eCopy in accordance with the standards provided by this guidance for the submission to be processed and accepted for review by FDA, unless they have been identified as being exempted. Submissions submitted without an eCopy and eCopy submissions that do not meet the standards provided in this guidance will be placed on hold until a valid eCopy is submitted to FDA and verified to meet the standards, unless a waiver or exemption has been granted. While the submission is on hold, the review clock will not begin.
In Section 745A(b), Congress granted explicit statutory authorization to FDA to implement the statutory eCopy requirement by providing standards, criteria for waivers, and exemptions in guidance. Accordingly, to the extent that this document provides such requirements under section 745A(b) of the FD&C Act (i.e., standards, criteria for waivers, and exemptions), indicated by the use of the words must or required, this document is not subject to the usual restrictions in FDA’s good guidance practice (GGP) regulations, such as the requirement that guidances not establish legally enforceable responsibilities. See 21 CFR 10.115(d).
However, this document also provides guidance on FDA’s interpretation of the statutory eCopy requirement and the Agency’s current thinking on the best means for implementing other aspects of the eCopy program. Therefore, to the extent that this document includes provisions that are not “standards,” “criteria for waivers,” or “exemptions” under section 745A(b)(2), this document does not create or confer any rights for or on any person and does not operate to bind FDA or the public, but does represent the Agency’s current thinking on this topic. The use of the word should in such parts of this guidance means that something is suggested or recommended, but not required. You can use an alternative approach if the approach satisfies the requirements of the applicable statutes and regulations. If you want to discuss an alternative approach, contact the FDA staff responsible for implementing this guidance. If you cannot identify the appropriate FDA staff, call the appropriate number listed on the title page of this guidance.
To comply with the GGP regulations and make sure that regulated entities and the public understand that guidance documents are nonbinding, FDA guidances ordinarily contain standard language explaining that guidances should be viewed only as recommendations unless specific regulatory or statutory requirements are cited. FDA is not including this standard language in this guidance because it is not an accurate description of all of the effects of this guidance. This guidance contains both binding and nonbinding provisions. Insofar as this guidance provides “standards,” “criteria for waivers,” and “exemptions” pursuant to section 745A(b) of the FD&C Act, it will have binding effect.
The eCopy Program is not intended to impact (reduce or increase) the type or amount of data the applicant1 includes in a submission to support clearance or approval. Please refer to other FDA device or program-specific guidance documents from CDRH (http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/default.htm) and CBER http://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/General/ucm214106.htm) for the appropriate contents for submissions.
An electronic copy (eCopy) is defined as an exact duplicate of the paper submission, created and submitted on a compact disc (CD), digital video disc (DVD), or a flash drive, accompanied by a copy of the signed2 cover letter and the complete paper submission.3
Note that inclusion of individual CDs for certain sections of the submission within the paper copy (e.g., a CD of the data line listings added to a sleeve in Attachment 3) will no longer be accepted because such CDs will not meet the requirements for a valid eCopy outlined in this guidance. Any CD provided must be an eCopy as defined above for the submission to be accepted.
While an eCopy is defined as an exact duplicate of the paper copy, there are limited cases in which differences between the eCopy and the paper copy may be justified because a paper copy is not practical or appropriate for analysis purposes (e.g., raw data and statistical analysis programs,4 data line listings to facilitate a bioresearch monitoring review) or is not feasible (e.g., videos, x-rays). The critical attribute of an eCopy is that it must include in electronic form all data required for that submission type.5 In other words, the eCopy must include all of the required information for FDA review, whereas the paper copy can include a placeholder cross-referencing the location of certain information in the eCopy.
The cover letter must contain the eCopy statement described in Attachment 1 and describe any differences between the paper version and the eCopy. If information is included in the eCopy that is not in the paper copy, then the paper copy must have a placeholder to redirect the reviewer (e.g., a piece of paper with a statement directing the reviewer to a particular section in the eCopy).
FDA will consider the eCopy (with the cover letter) loaded into the appropriate Center’s official document repository to be the official record. Any undisclosed differences between the eCopy and the paper version may need to be rectified and could delay the review of the submission.
Section 745A(b) of the FD&C Act, as added by section 1136 of FDASIA, requires an eCopy for the following submission types:
Premarket notification submissions (510(k)s), including third party 510(k)s;
Evaluation of automatic class III designation petitions (de novos);
Premarket approval applications (PMAs), including Transitional PMAs;6
Modular PMAs;
Product development protocols (PDPs);
Investigational device exemptions (IDEs) [see partial exemption below];
Humanitarian device exemptions (HDEs), including Humanitarian Use Device designation requests (HUDs);
Emergency Use Authorizations (EUAs)7 [see exemption below];
Certain investigational new drug applications (INDs);8
Certain biologics license applications (BLAs);9 and
Pre-Submissions.10
eCopies for all subsequent submissions to an original submission, including amendments (including add-to-files, CLIA categorization add-to-files, both of which are types of amendments), supplements, and reports11 to the submission types identified above would also be required even if the original was submitted to FDA prior to implementation of the eCopy requirements.
The size of the submission is irrelevant. Whether it is a single-page submission (i.e., the cover letter is the only content) or a multi-volume submission, the eCopy requirements apply.
Because responses to deficiency letters are required to be formally submitted to CDRH’s or CBER’s Document Control Center12 (DCC) to be logged in as an amendment or supplement, depending on the submission type, these responses are subject to the eCopy requirements.
Please note that eCopy requirements do not apply to information obtained during the Interactive Review process (via email, phone, and/or fax) once a submission is under review. However, should an applicant submit a response to an Interactive Review request to CDRH’s or CBER’s DCC (which should only occur if the size of the response makes communication by email or fax infeasible), it will be logged in and subject to the eCopy requirements.
Exemptions
Above, FDA identified the submission types cited in the legislation as being subject to the eCopy requirements. However, the legislation also allows for FDA to set forth criteria for exemptions from eCopy requirements. Accordingly, due to their potential urgent nature of the following types of submissions, FDA considers these to be exempt from the requirement for an eCopy:
two specific types of IDEs - compassionate use IDE submissions and emergency use IDE submissions;13 and
all EUAs.
Although these submission types do not require eCopies as per this exemption, FDA encourages you to submit eCopies of these submissions, when feasible, in order to facilitate the review process. If you choose to submit an eCopy, it must meet the standards outlined in Attachment 1.14
Waivers
FDA believes that, given the widespread availability of software to enable the creation of an acceptable eCopy at little to no cost, all applicants should have the ability to provide an eCopy. Therefore, at this time, FDA does not anticipate the need for waivers, except as described in Section 8.
Are there other submission types not subject to the eCopy legislation for which eCopies may be submitted?
Although not subject to the eCopy requirements of Section 745A(b) of the FD&C Act, FDA also accepts and strongly encourages you to submit eCopies for:
Master Access Files (MAFs);
513(g) Requests for Information (513(g)s); and
Clinical Laboratory Improvement Act (CLIA) Categorization – Exempt Device submissions (“X” files).
eCopies for these three submission types are voluntary; however, if you choose to submit an eCopy, it must meet the standards outlined in Attachment 1.
The eCopy Program does not change the overall number of copies to submit to FDA. Table 1 below provides the number of copies associated with each submission type for which an eCopy is either required or voluntary.
Table 1 – Number of Copies for Submission
Submission Type |
Required or Voluntary eCopy |
Total Number of Copies |
510(k)s |
Required |
215 |
Third Party 510(k)s |
Required |
2Error: Reference source not found |
De Novos |
Required |
2 |
PMAs, including Transitional PMAs |
|
|
|
Required |
616 |
|
Required |
6Error: Reference source not found |
|
Required |
317 |
|
Required |
2 |
Modular PMAs |
Required |
3 |
PDPs |
Required |
See PMAs for corresponding type |
IDEs |
|
|
|
Voluntary |
318 |
|
Voluntary |
3Error: Reference source not found |
|
Required |
3Error: Reference source not found |
HDEs, including HUDs |
|
|
|
Required |
See PMAs for corresponding type19 |
|
Required |
220 |
EUAs |
Voluntary |
3 |
INDs |
Required |
321 |
BLAs |
Required |
3 |
Pre-Submissions |
Required |
3 |
MAFs |
Voluntary |
2 |
513(g)s |
Voluntary |
3 |
CLIA X Files |
Voluntary |
3 |
For those submission types for which an eCopy is required, an eCopy (with a signed cover letter and an adequate eCopy statement) will serve as one of the required number of copies for the applicable submission types. FDA will accept additional eCopies in lieu of additional paper copies as long as at least one paper copy is submitted along with the eCopy and the total number of required copies remains the same.
For submission types for which only two copies are required to be submitted, one must be an eCopy and the other must be a paper copy. For submission types requiring more than two copies, this policy would allow additional flexibility in the format of the application that is submitted. For example, for an original PMA, you would submit: (1) one eCopy and five paper copies; (2) five eCopies and one paper copy; or (3) any other combination that results in six total copies as long as there is at least one eCopy and one paper copy.22
You may choose to submit only paper copies for compassionate use IDE submissions, emergency use IDE submissions, EUAs, MAFs, 513(g)s, and CLIA X files. However, if you choose to submit an eCopy for one of these, see above for details.
Below are the processing steps for the submission and acceptance of an eCopy.
With regard to the standards for an eCopy submitted to FDA, please refer to Attachment 1. An eCopy that does not meet the standards in Attachment 1 will fail to be accepted by our loading system.
At this time, there is no FDA tool available to pre-validate an eCopy that has already been developed by an applicant. However, there is a new free eSubmitter-eCopies tool available on FDA’s website at http://www.fda.gov/ForIndustry/FDAeSubmitter/ucm317334.htm, which we strongly encourage applicants to use. Use of this tool is optional; however, one of the benefits of the tool is that it creates an eCopy in real-time that is consistent with the standards described in Attachment 1. Use of the eSubmitter-eCopies tool is intended to prevent delays in review of your submission due to the need to resolve technical issues.
Should you have any technical questions when generating your eCopy, please contact cdrhesub@cdrh.fda.gov prior to submission of the eCopy to FDA.
In the case that another party (e.g., law firm, consultant) submits a submission on behalf of an applicant, the eCopy must still meet the standards for an eCopy in order to be successfully processed, whether accomplished by you (the applicant) or the submitting party. While the applicant may or may not include their own cover letter as part of the eCopy, our standards require that the submitting party include a signed cover letter with an eCopy statement, as described in Attachment 1.
There are two distinct parties involved in the generation of a Third Party 510(k): (1) the Accredited Person and (2) the applicant. Each party is subject to the eCopy requirements. Accordingly, each party (i.e., the Accredited Person and the applicant) must provide their own:
eCopy (on a single CD, DVD, or flash drive) that meets the standards in Attachment 1; and
signed cover letter with an eCopy statement as described in Attachment 1.
Therefore, there will be two separate eCopies provided for a given Third Party 510(k). Given this, it is essential that each eCopy be clearly marked as belonging to the Accredited Person or the applicant.
FDA recognizes that an applicant may interact with the Accredited Person multiple times prior to the Third Party 510(k) being submitted to FDA. Regardless of the number of interactions prior to the Third Party 510(k) being submitted to FDA, the applicant’s eCopy must be on a single CD, DVD, or flash drive in order to be loaded by our software. This could be accomplished by, for example, organizing each round of interaction as different volumes in the eCopy (e.g., VOL_001_Original Review, VOL_002_Round 2 Review). The Accredited Person must also provide their eCopy on a single CD, DVD, or flash drive for the same reason.
Although the Accredited Person is FDA’s point-of-contact and is the party that will be sent the eCopy hold notification if there are any issues with either eCopy, each party is responsible for meeting the eCopy requirements.
For bundled PMA or HDE submissions, there should be one version of the cover letter and the eCopy that applies to all submissions in the bundle. There should not be different cover letters associated with each submission in the bundle as has been common with past submissions.
In addition to the cover letter being signed and having an adequate eCopy statement, it should include a list or table of all submissions that are part of the bundle. The list or table should specify the submission number, trade name, and, as applicable, the model number, of each device impacted by the change.
An eCopy is submitted simultaneously with the paper submission(s). First, attach the signed cover letter with the eCopy statement to your eCopy. Then attach this eCopy package to the paper submission(s) and send them to CDRH’s or CBER’s DCC. An eCopy that is sent to the DCC without a cover letter and accompanying paper submission(s) will be placed on eCopy hold.
If more than one eCopy is to be submitted, then a single cover letter for all eCopies is sufficient.
The determination as to whether or not an eCopy passes the loading process will be made at the same time the submission is received by FDA and logged into our database.
If an eCopy passes the loading process, the cover letter and eCopy contents will be loaded into the appropriate Center’s official submission repository.
If an eCopy fails the loading process (i.e., is rejected), we will notify you in writing (e.g., by letter, email, and/or fax) that your submission is on eCopy hold. The notification will describe the reasons for the eCopy failure and the logistics for submitting a replacement eCopy. It is important that you follow these directions to avoid delays in processing the replacement eCopy. The submission will be placed and remain on eCopy hold until a valid replacement eCopy is submitted to FDA and verified to meet the standards.23 If you do not provide a replacement eCopy within 180 days, the submission will be deleted from our database.
As stated above, you will receive a notification that states the specific reason(s) why your eCopy failed the loading process. The reasons cited may or may not include issues with your cover letter, such as lack of an adequate eCopy statement or a signature, as described in Attachment 1.
In response to the eCopy hold notification, you must provide: (1) a revised cover letter that states you are providing a replacement eCopy and (2) a replacement eCopy (CD, DVD, or flash drive).
The revised cover letter is required regardless of whether or not the eCopy failure was related to the cover letter, because the DCC needs to be able to date stamp the revised cover letter to record the receipt date of the replacement eCopy. Be sure that your revised cover letter includes a signature and an adequate eCopy statement.
When FDA receives a replacement eCopy, it is processed in the same manner as the initial eCopy. More specifically, a determination is made as to whether or not the replacement eCopy passes the loading process. If it does not, then the submission will be placed on eCopy hold again, and an eCopy hold notification will be issued to you.
Review of a submission will not begin until a valid eCopy has been received and, if applicable, the user fee has been paid.
Furthermore, if applicable for that submission type, acceptance and/or filing reviews will be conducted. Otherwise, the substantive review of the submission will begin.24
If you submited an eCopy for a submission type that did not require an eCopy and you received an eCopy hold letter, what are your options?
If you submitted an eCopy for a compassionate use IDE, emergency use IDE, EUA, MAF, 513(g), or CLIA X submission and it did not meet the standards in Attachment 1, your submission will be placed on eCopy hold. However, unlike the other submission types that require a valid eCopy, you have the option of responding to that eCopy hold notification with an additional paper copy in lieu of a replacement eCopy.
Yes, unless your submission is an entirely electronic submission exempted under this guidance, as described below. Upon implementation of the statutory requirement, all medical device submission types listed in Section 4 must be accompanied by an eCopy regardless of the Center in FDA in which the submission will be reviewed unless the requirement is waived or exempted. Accordingly, submissions for devices subject to review under the FD&C Act and submitted by filing paper copies with CBER’s DCC must be accompanied by an eCopy, except where exempted as described below.
While many submissions made to CBER are still in paper format and require submission of multiple copies, CBER is also currently able to receive and manage submissions that are entirely electronic.
Submissions for devices that are subject to licensure under the Public Health Service (PHS) Act, including biologics license applications and supplements, investigational new drug applications, and EUAs and pre-submissions for these devices, may be submitted as entirely electronic submissions as detailed in Sections 8b and 8c below. FDA will exempt such entirely electronic submissions from the eCopy requirement.
FDA additionally waives the eCopy requirement to submit paper copies of any entirely electronic submission made to CBER. Accordingly, entirely electronic submissions that comply with CBER guidance identified in Section 8c below do not need to be accompanied by paper copies.
Yes, and there are several advantages for both industry and for CBER staff when you choose to make submissions electronically.
The main advantage to you is in the financial savings that will likely result. The costs associated with printing, binding, labeling, and shipping multiple paper copies can be significant, especially for submissions that contain a great deal of supporting documentation. Likewise, we anticipate that FDA will recognize financial savings in that FDA avoids the costs associated with tracking, routing, and storing large amounts of paper when you choose to submit electronically.
Another advantage with the use of the electronic submission process is that all parties involved in the submission and review are referencing the same document – the electronic one. There is no question about whether the paper copy is an exact copy of the eCopy. Electronic submissions may also reduce the need for reviewers to request re-submission of previously submitted information due to an inability to read or interpret the information on the paper copy, as sometimes occurs when documents are photocopied.
CBER has several resources available to applicants who choose to submit electronic submissions as outlined in the document “Regulatory Submissions in Electronic Format for Biologic Products.” (http://www.fda.gov/BiologicsBloodVaccines/DevelopmentApprovalProcess/ucm163685.htm). Thus, specific details are available in the cited references and will not be repeated in this guidance.
For devices that are regulated under the PHS Act and require the submission of a BLA, consult the guidance document entitled “Providing Regulatory Submissions to the Center for Biologics Evaluation and Research (CBER) in Electronic Format - Biologics Marketing Applications” (http://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/General/UCM192413.pdf) for details on preparing your electronic submission. Note that certain sections of this guidance, for example, those on pharmacology and toxicology, are generally not pertinent to licensed devices.
For guidance on preparing electronic submissions for other device submissions (e.g., 510(k)s, PMAs) sent to CBER, please see “Guidance for Industry: Providing Regulatory Submissions in Electronic Format - General Considerations” (www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM072390.pdf) and “CBER SOPP 8110: Submission of Paper Regulatory Applications to CBER” (http://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/ProceduresSOPPs/ucm079467.htm), which includes information about providing electronic copies to CBER.
We are currently developing additional, updated guidance for other electronic submissions sent to CBER and have issued a revised, updated draft guidance document for comment entitled, “Draft Guidance for Industry: Providing Regulatory Submissions in Electronic Format-General Considerations” (http://www.fda.gov/RegulatoryInformation/Guidances/ucm124737.htm). This document will provide an additional resource for applicants preparing electronic submissions.
You can submit questions pertaining to the preparation of submissions in electronic format for submission to CBER at ESUBPREP@fda.hhs.gov.
You may also contact CBER at CBER.CDISC@fda.hhs.gov to discuss the potential for submission of data in CDISC format (http://www.fda.gov/BiologicsBloodVaccines/DevelopmentApprovalProcess/ucm209137.htm).
CBER will accept electronic submissions via electronic transmission (i.e., through the Electronic Submissions Gateway25,26) or on physical media through CBER’s Document Control Center.
Attachment 1 –Standards for eCopies
Below are the standards that are written into the FDA eCopy software coding. If an eCopy does not meet all of the required standards identified in Sections A through D below, then the eCopy will not pass FDA’s eCopy loading process. Sections E through G provide additional items to consider when developing an eCopy.
In addition to the required signature,27 to meet the standards of the eCopy program, the cover letter must also include one of the following eCopy statements:
the eCopy is an exact duplicate of the paper copy; or
the eCopy is an exact duplicate of the paper copy except [specify all differences].
We expect the vast majority of the submissions to include the first statement. However, as described in Section 3 above, for those submissions that contain certain clinical datasets, or other types of data/information that is not feasible or practical to put in paper form, the second statement would apply.
If you include the second eCopy statement, you must specify the differences between the eCopy and paper submissions. In addition, you must include a placeholder in the paper submission referring to the eCopy for that specific information (e.g., “The eCopy is an exact duplicate of the paper copy except that data line listings were only provided in the eCopy. A placeholder was provided in Volume 4 of the paper copy referring back to the eCopy for that information.”).
Please also note that it is FDA’s preference that responses to deficiencies identified during submission review not be incorporated into the cover letter, for ease of processing and to minimize the paper scanning involved with the cover letter. Instead, please incorporate your responses into the main body of your submission.
The structure of an eCopy is highly dependent on the overall size of the submission and can be organized as a volume based or non-volume based submission as described below.
Volume-based eCopy
A volume-based eCopy is generally recommended for large or complex submissions so that the eCopy structure matches that of the paper version in order to facilitate the review of the submission. This eCopy structure includes volumes (i.e., folders) at the root level. Each volume, in turn, includes one or more PDF files.
A naming convention for the volumes is required in order to assure that the system can create a sort order of the folders that matches that of your paper submission. Each volume must have the following naming convention:
VOL_XXX (e.g., VOL_001); or
VOL_XXX_Descriptive Name (e.g., VOL_001_Patient Study Data).
The volume numbering must be non-repeating, consecutive numbers starting with VOL_001. The limit for volumes is 999.
Descriptive names for volumes are optional. However, if a descriptive name is used for a volume, the volume name should be descriptive of its content and meaningful to the reviewer. The descriptive name can be up to 250 characters but must not contain special characters (e.g., tilde (~), asterisk (*), forward slash (/), backward slash (\), colon (:), question mark (?), single quotation mark (‘), double quotation mark (“), less than sign (<), greater than sign (>) or vertical bar (|)).
If this volume naming convention is not followed, the eCopy will fail the loading process.
A slight variation of a volume-based eCopy structure includes both volumes and PDF files at the root level. This structure commonly occurs when an applicant adds a PDF of the cover letter at the root level with all other PDFs organized under multiple volumes.
NO SUBFOLDERS: Under this eCopy structure, you must avoid placing any subfolders under a volume or the eCopy will fail the loading process.
Non-volume-based eCopy
A non-volume based eCopy is generally recommended only for small submissions. This eCopy structure includes one or more PDFs at the root level.
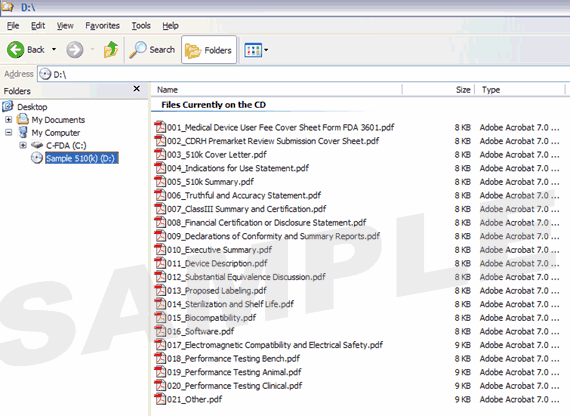
Figure 1 provides an example of a volume-based IDE. Figure 2 provides an example of a non-volume-based 510(k) submission that contains multiple PDF files.
Figure 1: Investigational Device Exemption (IDE) with Volumes
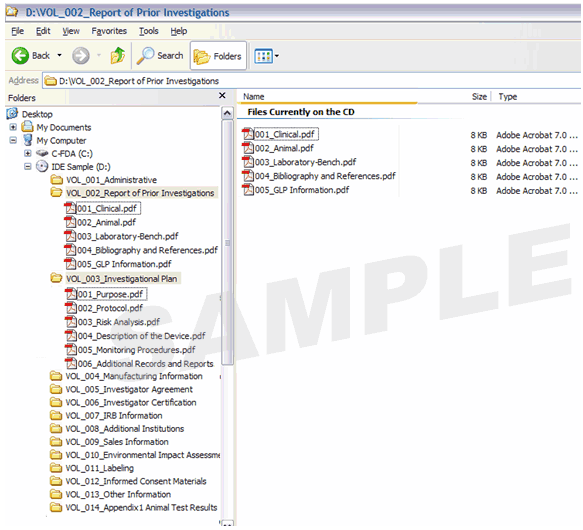
Figure 2: 510(k) Submission without Volumes
Whether you choose a volume or non-volume-based eCopy structure, PDF files are the primary file format used for an eCopy. (See Section D for how to add non-PDF files to an eCopy.) A PDF file ensures that no inadvertent changes occur to the submission and ensures that what a reviewer sees on the screen is the same as what has been submitted on paper. Below are the requirements for PDF files. If you do not follow them, your eCopy will fail the loading process.
Adobe Acrobat PDF Version 10.0 or Below
Only eCopies submitted using Adobe Acrobat 10.0 or below will be accepted.28 If you have a new version of Adobe Acrobat greater than 10.0, you must save the PDF as a reduced size PDF or the eCopy will fail the loading process.
No Attachments
eCopies submitted with attachments to the PDF files will not be accepted. Attachments must be removed before submittal. Many Acrobat PDF files created by a third party vendor, such as journal articles, user guides, and product labels, contain attachments which are embedded during the creation process. For example, see Figure 3 below, in which an attachment named “…joboptions” has been included. To detect attachments, open the Acrobat PDF and click the paper-clip icon in the lower left-hand corner of the Navigation Panel as shown in Figure 3 below.
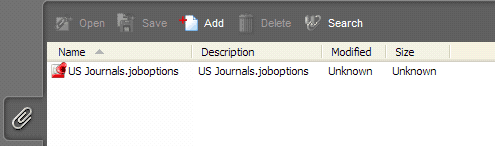
To remove the attachment:
contact the vendor directly and ask that they create the Acrobat PDF without appending any attachments like the one shown above; or
print the file selecting Adobe PDF as the Printer Name. The file will be printed to a new Acrobat PDF file without the attachment.
Note that manually deleting this attachment, rather than following the steps above, may not delete the attachment attribute from the file entirely.
PDF files with security settings will not be accepted. PDF files are stored as original documents and will not be altered from their original form. Remove any security settings, including read-only and password protection used on the files.
FDA recognizes that some of the forms on our website have password protection. We are currently replacing some of the primary forms used for premarket submissions with versions that do not include password protection. However, for FDA forms that still have password protection, your current options are to: (1) complete the form, print it, and scan it in or (2) save it in a zip file under the MISC FILES folder as described in Section D below.
A naming convention is required in order to assure that the loading system can create a sort order of PDF files, whether or not part of a volume-based submission, which will match that of your paper submission. You must use the following naming convention for all PDF files, whether part of a volume-based or non-volume based eCopy:
XXX_Descriptive Name (e.g., 001_Cover Letter, 002_MDUFA Form, 003_Table of Contents).
The PDF file numbering must be non-repeating, consecutive 3-digit numbers starting with 001. This naming convention applies even if there is only a single PDF file.
Keep in mind that if you have a volume-based submission, you need to start over with the numbering of the PDFs within each volume, as shown in Figure 1 above.
The descriptive file name can be up to 250 characters but must not contain special characters (e.g., tilde (~), asterisk (*), forward slash (/), backward slash (\), colon (:), question mark (?), single quotation marks (‘), double quotation marks (“), less than sign (<), greater than sign (>) or vertical bar (|)). When possible, the file name should be descriptive of its content and meaningful to the reviewers.
PDF File Size Limited to 50MB or Below
While there is no limitation on the total size of a submission, each PDF file must be limited to 50MB or smaller.
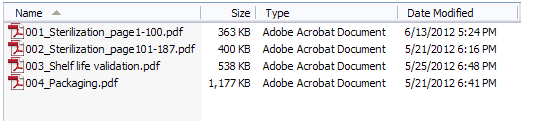
If a file size is greater than 50MB, then you must split the contents into multiple files and use the next file number in the naming sequence, as shown in Figure 4 below. If you must split your content, we recommend you name the files in a way that clearly reflects that. One way is to incorporate page numbers as shown in Figure 4.
Figure 4: Example of a PMA submission with page numbers in the file names
Section B describes how both a volume and non-volume-based eCopy include PDF files. However, an eCopy may also include non-PDF files. This is achieved by the inclusion of a “STATISTICAL DATA” and/or “MISC FILES” folder at the root level of the eCopy, regardless of whether it is a volume or non-volume based eCopy.
These two folders are optional; however, if you choose to incorporate one or both of them, the requirements are as follows:
spell the folder name precisely (i.e., STATISTICAL DATA, MISC FILES), although not case sensitive;
put the folder at the root level of the eCopy; and
include all content (see Parts 1 and 2 below) in one or more zip file(s) under the folder.
If you do not follow these requirements, your eCopy will fail the loading process.
Note that there are no limitations/restrictions on the naming convention for a zip file(s) or any of the content that you add to the zip file(s). In addition, there is no size limit for the zip files; however, it is recommended that you limit the size to 50MB or less in order to prevent potential problems, such as time delays when uploading the files.
Below describes the type of information appropriate as part of a “STATISTICAL DATA” or “MISC FILES” folder.
“STATISTICAL DATA” folder
Statistical information, including metadata29 and data line listings, may be included in the eCopy in their native formats, such as, but not limited to: SAS; XPORT; XML; SGML; S-Plus; R files; ASCII; Molfiles; and Excel. There are no restrictions on the native format.
“MISC FILES” folder
Some submissions may require miscellaneous files (e.g., videos, x-rays, machine readable software source code) that cannot be submitted (or should not be submitted) in PDF format and are not statistical in nature. These miscellaneous files may be included in the eCopy under the MISC FILES folder in their native formats, such as, but not limited to: .gif; .tif; .jpg; .avi; .mpeg; .wmv; and .txt. There are no restrictions on the native format.
In addition, for the purposes of streamlining the review process, FDA encourages you to also include, under the MISC FILES folder, Word versions of certain documents or pieces of information that were also provided in the main body of the eCopy as PDFs. In other words, include the PDF version in the main body of the eCopy as part of the volume or non-volume based eCopy structure and include a Word version in the MISC FILES folder to assist the reviewer. Documents such as those listed below are commonly requested via Interactive Review to enable FDA feedback to the applicant and/or completion of the review. Inclusion of these documents within the MISC FILES folder in an eCopy can help to minimize potential delays during the substantive review of the submission. Suggested documents include, as applicable:
Labeling for any submission [preferably with each piece (e.g., physician labeling, patient labeling, operators manual) as a separate file];
Predicate device comparison table for 510(k)s;
510(k) Summary;
Summary of Safety and Effectiveness Data (SSED) for PMAs; and
Summary of Safety and Probable Benefit (SSPB) for HDEs.
As stated above, there are no limitations/restrictions on the naming convention for any files you add to the zip files for the MISC FILES folder. However, when it comes to the Word documents included in the MISC FILES folder, it is recommended that you use a naming convention similar to the PDF equivalent files so that reviewers can easily make the correlation.
As stated in Section C.3. above, FDA recognizes that some of the forms on our website have password protection. One option to add a FDA form with password protection to an eCopy is to add it to a zip file under the MISC FILES folder.
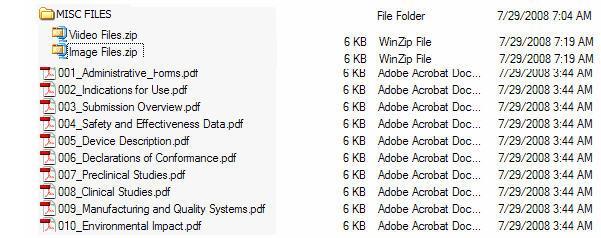
Figure 5 provides an example of an eCopy with a STATISTICAL DATA folder. Figure 6 provides an example of an eCopy with a MISC FILES folder. Both reflect zip file(s) directly under the folder.
Figure 5: eCopy with STATISTICAL DATA folder
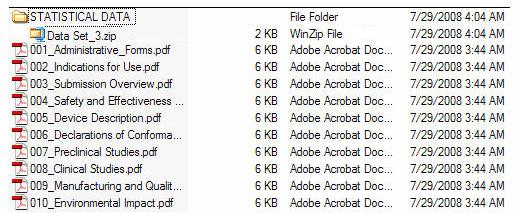
Figure 6: eCopy with MISC FILES folder
Bookmarks and hyperlinks within a single PDF file should be used to assist the reviewers in navigating through the content of the submission. If you use either bookmarks or hypertext links, consider the following:
Bookmarks
Bookmark references can be created for the heading of a section, subsection, or title of figures and tables within a single PDF file. In general, including a meaningful bookmark to the main table of contents for a submission or item is helpful, and will aid the reviewer in locating information and navigating the submission. When creating bookmarks, the magnification setting should be set to Inherit Zoom so that the destination page displays at the same magnification level that the reviewer is using for the rest of the document.
Hyperlinks
Hyperlinks are used to improve navigation through individual PDF documents and are encouraged. Hyperlinks can be designated by rectangles using thin lines or by blue text or you can use invisible rectangles for hypertext links in a table of contents to avoid obscuring text. Hyperlinks throughout the body of the document to supporting annotations, related sections, references, appendices, tables, or figures that are not located on the same page are helpful and improve navigation efficiency.
Hyperlinks to other sections within an individual PDF document are permitted and maintained within a given PDF file when added to a Center’s official document repository. However, hyperlinks from one PDF to another PDF file (or across other file types) are not supported and will not work at this time.
When creating your PDF from the source document (e.g., Word document), please consider the following:
If you use Adobe plug-ins to create PDF files and/or capture or display data, please note that there is a risk that information may not display correctly because reviewers may not have access to certain plug-ins to review content being displayed by a plug-in.
Fonts
Any documents displayed publicly by FDA (such as 510(k) Summaries and PMA Summaries of Safety and Effectiveness Data) must be in compliance with Section 508 of the Rehabilitation Act (29 U.S.C. 794d), as amended by the Workforce Investment Act of 1998 (P.L. 105-220), August 7, 1998. In order to ensure Section 508 compliance and for easier reading by FDA staff, one of the following fonts should be used in your source document: Times New Roman; Verdana; Arial; Tahoma; or Helvetica. You should avoid using customized fonts and multiple fonts within the same document.
We recommend the use of a black font color. Blue font may be used for hypertext links. If a font color other than black is used, avoid light colors that do not print well on grayscale printers. It is advised that you test the color reproduction prior to submission by printing sample pages from the document using a grayscale printer. In addition to font colors, keep formatting simple in tables.
The applicant should create all PDF versions directly from the source documents whenever feasible rather than by scanning. PDF documents produced by scanning paper documents are far inferior to those produced from an electronic source document, such as Word, and, thus, should be avoided if at all possible. Scanned documents, particularly tables and graphs, are more difficult to read and do not allow the reviewers to search, or copy and paste text for editing.
If scanning cannot be avoided, the following is highly recommended:
Perform optical character recognition (OCR) on all scanned documents so that the text is searchable. Check to see that the content has been correctly converted by: (1) highlighting an area of text and (2) searching for a word or phrase. If the word or phrase is not returned in the search, then the OCR did not recognize the text. FDA recognizes that OCR may not be feasible in some cases for documents with figures and images.
If the source document is only available on paper, it should be scanned at resolutions that will ensure the pages are legible both on the computer screen and when printed. At the same time, remember to limit the file size to 50MB or less. We recommend scanning at a resolution of 300 dots per inch (dpi) to balance legibility and file size. We discourage the use of grayscale or color because of file size. After scanning, avoid re-sampling to a lower resolution.
When creating PDF files containing images, you should not resample images. Resampling does not preserve all of the pixels in the original. For photographs, the image should be obtained with a resolution of 600 dpi. If black and white photos are submitted, consider 8-bit gray scale images. If color photos are submitted, consider 24-bit RGB Color Model images. A captured image should not be subjected to non-uniform scaling (e.g., sizing).
Files with scanned images and photographs tend to be large in file size, so be careful not to exceed 50MB for a single file or your eCopy will fail the loading process.
Paper documents containing handwritten notes must be scanned at 300 dpi. These handwritten notes should be made in black or blue ink for clarity.
1 For the purposes of this guidance, applicant includes “submitter,” “sponsor,” or “holder.”
2 The cover letter signature may be a wet (i.e., ink) signature or a valid digital signature. Please note that other forms submitted within an IDE or premarket submission (e.g., Truthful and Accurate Statement for 510(k)s) can also be signed with either a wet signature or digital signature.
3 An eCopy is not considered to be an electronic submission. For information on eSubmissions, refer to “FDA eSubmitter” (http://www.fda.gov/ForIndustry/FDAeSubmitter/default.htm) and “Regulatory Submissions in Electronic Format for Biologic Products” (http://www.fda.gov/BiologicsBloodVaccines/DevelopmentApprovalProcess/ucm163685.htm).
4 For information on electronically submitted data, refer to “Clinical Data for Premarket Submissions” (http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/HowtoMarketYourDevice/PremarketSubmissions/ucm136377.htm).
5 For example, the content requirements for a 510(k) submission are found in 21 CFR 870.87 and 807.92; those for original PMA submissions are found in 21 CFR 814.20.
6 This includes all PMA submission types, including, but not limited to, original PMAs, panel-track supplements, 180-day supplements, manufacturing site change supplements, 30-Day Notices, 135-Day Supplements, and post-approval study supplements, as well as changes in the correspondent or ownership and requests for extensions.
7 Refer to the guidance entitled, “Emergency Use Authorization of Medical Products” (http://www.fda.gov/RegulatoryInformation/Guidances/ucm125127.htm) for more information on EUAs.
8 Applicable only to those devices regulated by CBER that are also biologics under Section 351 of the Public Health Service (PHS) Act and that also require submission of an IND prior to submission of a BLA. Such devices are generally those intended for use in screening donated blood for transfusion transmissible diseases.
9 Applicable only to those devices regulated by CBER that are also biologics under Section 351 of the PHS Act, including those that do not require submission of an IND prior to the submission of the BLA. Such devices generally include those reagents used in determining donor/recipient compatibility in transfusion medicine in addition to those for use in screening blood for transfusion transmissible diseases.
10 Refer to the draft guidance entitled, “Medical Devices: The Pre-Submission Program and Meetings with FDA Staff” (http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm310375.htm).
11 Reports include all reports submitted to an applicable submission type, including annual/periodic and post-approval reports. Section 745A(b) of the FD&C Act does not apply to Medical Device Reports submitted under 21 CFR Part 803.
12 Refer to 21 CFR 807.90 for the DCC addresses for CDRH and CBER.
13 Please refer to CDRH’s device advice page entitled “IDE Early/Expanded Access” (http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/HowtoMarketYourDevice/InvestigationalDeviceExemptionIDE/ucm051345.htm#compassionateuse) and FDA’s “Guidance on IDE Policies and Procedures” (http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm080202.htm) for additional details on compassionate and emergency use IDE submissions.
14 If submission of an eCopy is not feasible, but there is pertinent electronic information, such as imaging data, to supplement the information in the paper copy, please contact the review branch for information on how to submit this information.
15 See 21 CFR 807.90(a)(3)(c).
16 See 21 CFR 814.20(b)(2).
17 See 21 CFR 814.39(c).
18 See 21 CFR 812.20(a)(3).
19 See 21 CFR 814.104(b)(4).
20 See 21 CFR 814.102(d).
21 See 21 CFR 312.23(d).
22 CDRH review staff should not request an additional eCopy or paper copy of a submission from the applicant. However, this should not be confused with requests for additional information made via the Interactive Review process during the review of a submission.
23 Do not confuse an “eCopy hold” with FDA decisions such as Refuse to Accept or Refuse to File. An eCopy hold takes place before a submission is subject to any review process. Once under review, if applicable for that submission type, acceptance and/or filing reviews will be performed. See also Section 7.i. of this guidance.
24 For more information, please see the guidances “FDA and Industry Actions on Premarket Notification (510(k)) Submissions: Effect on FDA Review Clock and Goals” (http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm089735.htm) and “Guidance for Industry and Food and Drug Administration Staff - FDA and Industry Actions on Premarket Approval Applications (PMAs): Effect on FDA Review Clock and Goals” (http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm089733.htm).
25 Refer to “Food and Drug Administration Electronic Submissions Gateway (Federal Register Notice) - 8/9/2006” (http://www.gpo.gov/fdsys/pkg/FR-2006-08-08/html/E6-12808.htm).
26 Refer to “Electronic Submissions Gateway” (http://www.fda.gov/ForIndustry/ElectronicSubmissionsGateway/default.htm).
27 You may also choose to include an electronic version of the cover letter in the eCopy; however, this should not be in lieu of the attached cover letter with a signature.
28 As we test and validate new versions of Adobe Acrobat, we will update this guidance accordingly.
29 Metadata includes data dictionaries and terminologies, formats, annotated case report forms, statistical analysis details, and any other information that contributes to understanding and using the data.
| File Type | application/msword |
| File Title | Attachment E CDRH Final Guidance Cover Sheet |
| Author | OHIP Info Systems |
| Last Modified By | Corbin, Abigail |
| File Modified | 2012-12-07 |
| File Created | 2012-12-07 |
© 2026 OMB.report | Privacy Policy