_SEED_Supp_Statement_B
_SEED_Supp_Statement_B.doc
The Study to Explore Early Development (SEED)
OMB: 0920-0741
B. Statistical Methods
B.1. Respondent Universe and Sampling Methods
SEED participants will be drawn from children born in and residing in the six study areas: the San Francisco Bay area, Denver metropolitan area, Philadelphia metropolitan area, Maryland, central North Carolina, and the Atlanta metropolitan area. Each SEED site selected 2-10 counties for its catchment area based on the following criteria: proximity to the study site, at least 30,000 live births collectively across counties, and geographic adjacency (i.e., counties must be contiguous). The sample frame for the cases and the NIC group, for each site, consists of all clinical and educational settings serving children with ASDs (both public and private) within the catchment areas. The sample frame for the subcohort is the birth certificates for all children born during the birth cohort, excluding those children who have died. Please see Appendix K (Ascertainment Methodology) for additional detail.
The study cohort consist of children:
Born from September 2003 – August 2006 (i.e., a 36-month birth interval) and
Born in and currently reside in the catchment area of each site during the study period, which is Winter 2007/08 - Summer 2012 (actual dates subject to change due to study implementation considerations).
The following criteria describe which children are potentially eligible for this study and justifications for these criteria:
Child is 24-60 months old at time of eligibility (birth date range of September 2003 to August 2006) This age range, which is younger than the age range seen in some other studies, was chosen in order to limit recall bias for events in pregnancy and early life as much as possible while still allowing diagnostic accuracy for ASD.
The child is between 30 – 68 months at completion of data collection components. This age range is appropriate for the instruments to be administered in the study.
Child was born in and currently resides in the study catchment area. The defined cohort (required by the case-cohort design) is to be ascertained from birth certificate data; current residence is required for ascertainment purposes and to allow for examinations and other in-person assessments of enrolled subjects.
Child currently resides with a knowledgeable caregiver. For purposes of the study, a knowledgeable caregiver is defined as a family member or other caregiver of legal age who has resided with and consistently been caring for the child since the child was 6 months of age or younger (based on self-report). This criterion is necessary in order to collect accurate information on early life events that may be risk factors for the development of autism.
Legal consent is obtainable.
Note. Any children with legal circumstances (e.g., legal adoption) preventing access to birth certificates/ legal consent are not eligible for the study. Foster children are excluded because, as wards of the state, it is difficult to obtain informed consent for them. Children who have been legally adopted will not be enrolled because birth certificate records are typically sealed. Birth certificates are essential in determining place of birth, one of the eligibility criteria for the study.
The knowledgeable caregiver is competent to communicate orally in English or Spanish. This limitation is necessary since the clinical study instruments to be administered are only available and validated in English, although the majority of the instruments are also validated in Spanish. Some sites may exclude Spanish-only speaking participants based on the percentage of Spanish-only speaking residents and other site specific factors.
Children who do not meet all of these criteria aren’t eligible to participate.
Identification of Potential Case and NIC Children
Potential cases and NIC children are cohort children identified by the study as having a suspected ASD or other selected neurodevelopmental impairments (please refer to ICD-9 codes/Part B School Eligibility Criteria [Appendix L] for list of diagnoses). For the purposes of this study, ASD includes Autistic Disorder, Pervasive Developmental Disorder-Not Otherwise Specified, and Asperger’s Syndrome. Potential cases and NIC children are identified through sources serving or evaluating children with developmental problems. Sources for potential cases and NIC children may include Part C and Part B agencies, special education programs, state autism registries, hospitals, and clinics. These types of data sources were chosen based on experience with CDC autism surveillance activities. Each SEED site obtained IRB approvals from their appropriate institutions and, if necessary, written agreements from their local sources. Site-specific case-ascertainment procedures, including definitions of Part C and Part B agencies, are detailed in SEED Case, Comparison Group, and Subcohort Ascertainment Methodology (Appendix K).
The first step was to identify potential case and NIC children who: a) have received an ASD diagnosis, b) meet specific ASD or ASD-related exceptionality criteria, or c) have received a diagnosis of one or more select conditions associated with ASD. Please see Appendix L (ICD-9 Codes/Part B School Eligibility Criteria) for specific eligibilities. In addition to having at least one of the criteria above, the child must meet all eligibility criteria listed in Section B.1.
Note that the criteria described above for children with suspected ASD are quite broad and are not limited to children with a previous autism diagnosis or autism exceptionality for early intervention/special education. This broad diagnostic net for possible cases ensures that young children with suspected ASD (i.e., young children without a formal diagnosis of ASD) are identified. However, it also identified many children who may not have ASD. Therefore, SEED sites use an ASD screening instrument to assign participants into the potential case group or the NIC group. All of the respondents who screen into the potential case group are invited to enroll in the study. However, since the study analysis plan requires a one-to-one case to control ratio, those respondents who screen into the NIC group may or may not be invited to enroll. At each SEED site, a sample of NIC children is drawn from all NIC children across the catchment area.
The purpose of the ASD screen is to identify those children in the broad net who are most likely to have ASD. The screening process consists of administering the Social Communication Questionnaire (SCQ, Appendix J). The SCQ is a 5 to 10 minute screening interview completed by the child’s primary caregiver. It is normed as a screening instrument for ASD in children 4 years and older.
Any child who has an ASD diagnosis or is receiving autism services from a public school (indicated in general by a code for autism special education services) receives a full diagnostic assessment and be included as a case if they meet study criteria. The main purpose for the SCQ screen in this study is to identify children who might meet case
criteria for SEED but who do not meet the aforementioned criteria. This group of children will principally be drawn from a pool of children receiving special education
services other than for ASD and children with other neurodevelopmental ICD-9 diagnoses (see Appendix L).
The SCQ is the considered to be the best parental screening instrument for this study. Although the published SCQ is validated on children 4 years of age and older, there has been considerable work recently investigating the performance of the SCQ (Appendix J) in children younger than age 4 (Wiggins, L., Bakeman, R., Adamson, L., & Robins, D., in press; Hanson, Sullivan, Ware, Lord, & Thurm, 2002; Corsello, Cook, & Leventhal, 2003; Corsello, Anderson, Qui, Risi, & Lord, 2004; Corsello, Lord, Hus, & Qui, 2005; Eaves, Wingert, Ho, & Mickelson, 2006; Eaves, Wingert, & Ho, 2006; Baird, et al., 2006), There has also been a study conducted by the MD SEED site in preparation for SEED (Lee, David, Rusyniak, Landa, & Newschaffer, in press). Lee et al (in press) found that the sensitivity and specificity of the SCQ is improved at a reduced cut-off score in younger age cohorts. Likewise, Wiggins et al. (in press) found that sensitivity and specificity improved at a cut-off score of 13 in children younger than 4 years of age; sensitivity and specificity were maximized at a cut-off score of 11. Finally, Allen et al. (Allen, Silove, Williams, & Hutchins, 2006) reported that when using a cut-off score of 11, sensitivity and specificity of the SCQ was 93% and 58% for children aged 2-6 years, and 100% and 62% for children aged 3-5 years.
Thus, a cutoff score of 11 is employed for this study. If the child scores > 11, ASD is considered indicated (i.e., positive screen). If the child scores <11, ASD is considered not indicated (i.e., negative screen).
In order to obtain a systematic sample representing a range of diagnoses (for the NIC group, and a range of ages (for both the NIC and case group), it is important to try to obtain the diagnosis of each potential participant prior to contact. However, some local sources are not able to release the diagnosis to SEED investigators without individual consent. Thus, the recruitment process and contact of potential cases and NIC children varies among sources within each study site. Two recruitment scenarios have been developed to address these differences prior to first participant contact: one for sources that are able to release the diagnosis/exceptionalities of potential participants and one for sources not able to release diagnoses/exceptionalities without consent. The recruitment scenarios are discussed below and shown in Figures 3 and 4.
a. Sources with Diagnosis Known to Investigators Prior to Initial Subject Contact
Figure 3 describes the recruitment process for local sources that releases information about the diagnosis/exceptionality of the children to SEED investigators without consent (i.e., prior to first contact with a potential participant). Based on the released information from these sources, subjects are identified for recruitment following the ASD screening process (SCQ screen):
Children with a previous ASD diagnosis/exceptionality are contacted, screened for eligibility, administered the SCQ, and then enrolled in the study.
Children identified by the broad diagnostic net who do not have a previous ASD diagnosis/exceptionality, a systematic sample representing a range of diagnoses and ages found in the database are used to identify which children will be invited to participate following the eligibility and autism screening steps. The proportion of the children identified by the broad diagnostic net criteria and subsequently sampled varies among sources depending on the site-specific requirements for subject contact (see Appendix K). At sites where consent is required before eligibility screening, the sample may be up to 100% of those consenting to eligibility screening if the proportion of subjects who are contacted and consent to the eligibility screen is low. The sample proportion may be lower than 100% at sites where consent is not required before contacting for the eligibility screening. Thus, the sample proportion is adjusted by each SEED site to ensure the target enrollment is met. Each child’s identified legal guardian is contacted to determine if the child meets the study eligibility criteria.
As an illustration of this approach, the GA SEED site (CDC) recruitment process is summarized as follows (details in Appendix K): 1) Following procedures established as part of CDC ASD surveillance, sources are requested to provide lists of children who meet the cohort eligibility and broad diagnostic net criteria. Based on the diagnostic/exceptionality information released by the sources children with a previous ASD diagnosis/exceptionality are contacted, screened for eligibility, administered the SCQ, and enrolled (Possible Case Group), 2) children without a previous ASD diagnosis/exceptionality are contacted, screened for eligibility, administered the SCQ, and enrolled in either the Possible Case or NIC groups based on the results of the SCQ screener. For the NIC group, the sample proportion to be contacted are adjusted to produce an approximate 1:1 ratio of case-NIC children.

b. Sources with Diagnosis Unknown to Investigators Prior to Initial Subject Contact
Figure 4. (p. 63) describes the recruitment process for local sources that are unable to release the specific diagnosis type/exceptionality to SEED investigators without consent from the potential participant. These sources have a slightly different process for
determining their potential case and NIC groups. Sites with this type of local sources go through the additional step of obtaining consent to contact prior to ASD screening and determining the eligibility of the potential participant.

c. Self Referrals
Parents, or legal guardians, who contact the study site, have a child with a previous ASD or ASD-related diagnosis or concerns that their child might have an undiagnosed ASD or ASD-related disorder, and are interested in having their child participate, are considered Self-Referrals. Self-referred children without documentation from a health care provider or other appropriate source of an ASD or ASD-related condition, however, will be ineligible for enrollment as a Self-Referral. Self-Referral procedures are detailed in Appendix K. Children in this group who have documentation of ASD or ASD-related criteria (Enrollment Packet, Appendix E), and meet all study eligibility criteria, will be enrolled and proceed through the study as a member of the Broad Net, as shown in Figures 3 or 4 (depending on the site-specific recruitment process). Regardless of the results of the screening, parents who express concern about their child’s development are given referral information for further developmental evaluation.
Identification of Potential Subcohort Children
Subcohort children are identified from birth certificates on the basis of birth date range and residence in the catchment area at the time of birth. In addition to IRB approval, most sites require approval from the State Registrar or Vital Records Department to obtain files containing personal identifiers. Once approvals are obtained, potential subcohort member children are randomly selected from among all cohort children. When possible, sites link birth records to state death certificate files to remove any children from the contact list who are deceased (Site-specific information on subcohort ascertainment is provided in Appendix K). The current residence in the study catchment area also needs to be established.
Based on these steps, contact takes place with each child’s identified legal guardian to obtain consent and to determine if the child meets the study eligibility criteria.
If the legal guardian consents to have the child participate in the study and the child meets the eligibility criteria, then the child is enrolled as a subcohort member as shown in Figure 5 (p. 65). All children enrolled in the subcohort receive the SCQ for the ASD screen. Children whose SCQ results indicate that the child is likely to have ASD are invited for a clinic visit to undergo a developmental evaluation following the data collection of the Case group. This evaluation includes autism specific assessments.
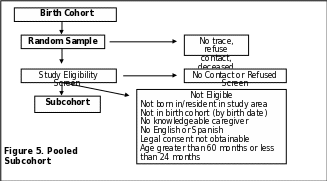
Final Subject Group Classification
1) ASD: ASD case confirmation is established by the current state-of-the-art methods by administering the Autism Diagnostic Interview (ADI-R, Appendix F.1) to the primary caregiver, and the appropriate module of the Autism Diagnostic Observation Schedule (ADOS, Appendix G.1) to the child. In some instances, (e.g. refusal, loss to follow-up) these subjects may not complete the study’s developmental evaluation and, thus, retain their initial assignment as Possible Case group member for analysis purposes. Consequently, analyses as outlined in A.16 may be performed separately on subjects classified on the basis of the developmental evaluation and subjects classified on the basis of the ASD screen.
2) NIC: A child is considered a member of the NIC when they have documentation of a condition listed in Appendix L indicative of a neurologically impaired developmental disability diagnosis and receive a score on the SCQ indicating that autism is unlikely.
Possible Case children who do not meet ASD criteria based on the complete developmental evaluation are also designated a member of the NIC group.
3) Sub-cohort: A child that is sampled from the birth cohort, and meets study eligibility criteria, remains a member in the sub-cohort comparison group. If a member of the sub-cohort group has a positive screen on the SCQ, they also complete the same evaluation as that for a possible case. If they meet case criteria based on the developmental evaluation they will be included in the final case group.
Sample Sizes and Response Rates
The proposed cohort size for the study (approximately 580,000 births combined across all 6 study sites, based on a range of 33,000-52,000 births per year in each study site catchment area) and therefore, the anticipated number of individuals in the case and comparison groups, was partly determined by time and resource considerations. Details on expected case sample size and study power are provided in B2. In short, our target final sample size – i.e., the number of enrolled children with complete data collection - is about 650 children in each of the three subject groups (Case, NIC, sub-cohort), or about 1950 children in total. Based on the experience of the CHARGE Study regarding the percentages of cases and sub-cohort members who were eligible for the study, agreed to participate, and completed data collection (CHARGE investigators, personal communication; Hertz-Picciotto et al 2006), as well as Georgia SEED experience (Georgia SEED is the CDC SEED study site) during the first 21 months of implementation, the following expected samples sizes (all sites combined) at different phases of the study are provided below (we expect the corresponding samples sizes for the NIC group to fall within the range of the ASD and sub-cohort estimates, with rates of participation assumed to be more similar to the sub-cohort group than ASD group):
Sub-cohort: 8,506 1 x 32% confirmed contact = 2,722 x 67% eligible = 1,8243 x 51% participation = 9304 x 70% complete data = 6505
NIC: 4,510 2 x 49% confirmed contact = 2,210 x 69% eligible =1,525 3 x 61% participation = 9304 x 70% complete data = 6505
ASD: 2,622 2 x 55% confirmed contact = 1,442 x 86% eligible =1,240 3 x 75% participation = 9304 x 70% complete data = 6505
1 random selection of potential sub-cohort children from birth cohort
2 number of potential ASD cases or NIC children in birth cohort; for ASD cases, this number is derived from the number of potential cases in the cohort (with a previous diagnosis or true positives on the ASD screen) based on a conservative estimate of the ASD rate of 4.5 per 1000 among all cohort births – we expect the prevalence rate to be at least this value based on recent ASD prevalence estimates in older children; for NIC, this number corresponds to the minimum number of children with a broad net diagnosis who screen negative on the ASD screen.
3 number of children (families) determined eligible for enrollment after contact
4 number of enrolled children (families)
5 number of enrolled children (families) with complete data collection
B2.Procedures for the Collection of Information
Sample size and Study Power Estimation
The proposed cohort size for the study (approximately 580,000 births combined across all 6 study sites, based on a range of 33,000-52,000 births per year in each study site catchment area) and therefore, the anticipated number of cases and comparison group children, was partly determined by time and resource considerations. Further, few data are available to estimate anticipated effect sizes for particular exposure-outcome relationships of interest. Given these constraints, we calculated what the minimum effect sizes (relative risk estimates) would be with the anticipated sample size.
The anticipated sample size was based on CDC-provided prevalence estimates from the Metropolitan Atlanta Developmental Disabilities Surveillance Project (MADDSP) corresponding to 3.4 ASD cases per 1,000 children ages 3-10 years in the population in 1996, as well as a published range in the literature of 2-10 per 1000). Because we are focusing on preschool children, we chose a conservative, minimum prevalence estimate of between 4.0-5.0 per 1000 children. Thus, with 580,000 births in the birth cohort across all study sites, we anticipate identifying about 2,622 ASD cases (based on a prevalence of 4.5 per 1000), including previously diagnosed children and SEED-identified ASD children. Based on the experience of the CHARGE Study regarding the percentages of cases who were contacted, were eligible for the study, agreed to participate, and completed data collection (CHARGE investigators, personal communication; Hertz-Picciotto et al 2006), as well as Georgia SEED experience (Georgia SEED is the CDC study site) during the first 21 months of implementation, the following expected final sample size (all sites combined) is:
ASD: 2,622 x 55% confirmed contact = 1,442 x 86% eligible = 1,240 x 75% participation = 930 x 70% complete data =
650 enrolled ASD children (families) with complete data collection.
We enroll subjects in each comparison group in a 1:1 ratio to cases. Although the study has two comparison groups, only one group is compared to the cases at a time.
SEED estimation of minimum detectable Relative Risk (RR) estimates (or minimum detectable Odds Ratios (OR)) was based on simple case-control approaches. NCSS PASS 6.0 software was used for all calculations.
A number of estimations were made to gauge the effect of different assumptions (case group size – all cases or stratified on specific ASD subgroups - and exposure prevalence rates) on min Odds Ratios (OR). For all estimates, conventional alpha and beta error tolerances were applied (0.05 and 0.20, respectively). The assumptions and range of min OR are tabulated below:
-
Size of ASD case subgroups of etiologic interest
Proportion of full case group
Corresponding Subgroup
20%
Complex autism
30%
Nonverbal; With Regression
40%
With MR
60%
No MR
70%
Verbal; Without Regression
80%
Essential autism
-
Select candidate exposure prevalence rates
Subcohort Exposure Prevalence Estimate
Corresponding Candidate Exposures
1%
ART, OCs
5%
Any infertility tx, maternal thyroid disorder hx, seizures, FraX
10%
Any maternal autoimmune hx, ADHD, GI dysfunction
20%
Pitocin, fever in pregnancy
50%
Any infection in pregnancy
S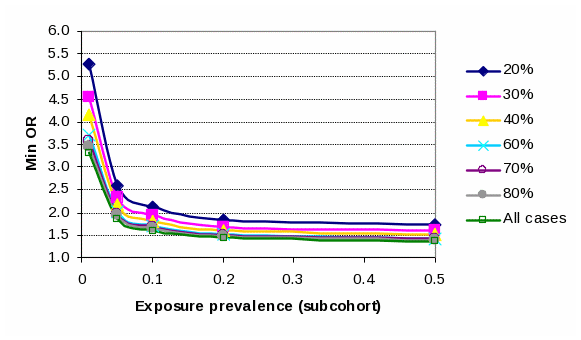 EED
CASE-CONTROL STUDY POWER ANALYSES
EED
CASE-CONTROL STUDY POWER ANALYSES
Case
subgroup

Minimum detectable OR (80% power, alpha error tolerance 5%) under alternate subcohort exposure prevalences and ASD case subgroup sizes |
|||||||
Subcohort Exposure |
Size of case subgroup as a % of total case group (650) |
||||||
Prevalence |
20% |
30% |
40% |
60% |
70% |
80% |
All cases |
1% |
5.28 |
4.55 |
4.15 |
3.71 |
3.57 |
3.46 |
3.31 |
5% |
2.60 |
2.33 |
2.18 |
2.02 |
1.97 |
1.93 |
1.87 |
10% |
2.12 |
1.93 |
1.82 |
1.71 |
1.67 |
1.65 |
1.61 |
20% |
1.84 |
1.69 |
1.61 |
1.52 |
1.50 |
1.48 |
1.45 |
50% |
1.72 |
1.59 |
1.52 |
1.43 |
1.41 |
1.39 |
1.37 |
Last, minimum detectable effects for interaction were calculated:
Interaction assessment:
Exposure 1 prevalence: 0.025, 0.05, 0.20, 0.35, 0.50; Exposure 2 prevalence: 0.05, 0.10, 0.25, 0.50
For the minimum detectable interaction odds ratio (assuming no stratification and a 1:1 case-control ratio), the values ranged from about 5.0 to 12.0 for combinations of the lowest prevalence rates for Exposure 1 with different Exposure 2 prevalence rates; they declined to about 2.0 to 3.5 for combinations of the highest prevalence rates for Exposure 1 with different Exposure 2 prevalence rates.
Considering the factors used in the above calculations (e.g., exposure prevalence rates, ASD subgroup size), the factors that are possibly modifiable by design to enhance study power are the case: control ratio and case group size. However, compared to a 1:1 case-control ratio, a 1:2 ratio had relatively small effects on the mdRR. Thus, it was determined that the greatest benefit would be gained by enhancing case group size by taking steps to increase participation and/or enhancing case ascertainment (e.g., the “Broad Net of Neurodevelopmentally Impaired Children”) from the proposed study cohort. An additional step, to be planned for the future (if funding levels permit), is to expand the study cohort and continue enrollment into a second phase of field data collection.
With regard to power for gene-environment interactions, we acknowledge that 650 cases/ sub-cohort members is on the lower end of desired sample sizes for tests of interaction and, if feasible, we would certainly advocate enrolling more subjects. However, such an ideal world is rarely possible, and our task is to use our expertise to balance “perfect study” intentions with the practical limitations of the real world in a way that retains meaningful information. After such an exercise, we decided upon our current approach as the best way to achieve that balance.
Our reasons include consideration of phenotype precision versus sample size and the current lack of agreement in the field about how to best address the multiplicity of testing when considering large-scale genotyping efforts. The first point has been a driving force. The power to detect a causal effect is driven not only by sample size, but also by the amount of error around each measurement used in an analysis. We believe that careful phenotyping provides much less misclassification and much greater precision than less costly protocols that would allow larger sampling. Further, the main concern regarding power for gene-environment interactions appears to stem from an assumption of genome-wide genotyping, or at least very large numbers of genotypes to be generated, and the need to correct for multiple tests, which reduces power estimates by requiring a very strict threshold for significance. While this is worthy of consideration, the genetic epidemiology field as a whole has not yet agreed upon an approach. Some researchers have promoted the use of a Bonferroni correction for all markers genotyped, although most agree that this is too conservative. Several would argue in fact that no correction is needed, and that biological plausibility and replication removes false positive findings. Others promote methods such as allowing a specified false discovery rate, or applying a Bayesian approach relying on priors for different sets of GxE tests. Each of these has merit, and would suggest a different “significance” threshold for calculations of power. Under the assumption that correction is irrelevant, we would have very high power estimates for our approach. Under the opposite extreme, using a Bonferroni correction for half a million tests, power estimates would be quite low. Most of the other approaches would yield results in between, but would suggest we have ample power to
detect moderate to large GxE effects. Given the various aims of this project, we feel this is the appropriate compromise of size versus precision for this study.
Procedures for the Collection of Information
Identification of case, NIC, and subcohort subjects is described in Section B.1. Once the children are identified for the study, an introductory letter (Appendix M.1), study brochure (Appendix M.2), pre-paid response card (Appendix M.3), and incentive valued at one dollar are mailed to the primary caregiver. This introductory packet is mailed from the study site (e.g., CDC) or collaborating local sources (e.g., Montgomery County Department of Education) depending on the agreements each site has with their various sources. All introductory packets are sent in English. For sites including Spanish speakers (California and Colorado), the materials are sent in both English and Spanish and the response card included in this packet asks the primary caregiver to indicate language preference, so that all future materials can be mailed in the appropriate language.
If no response card is received within two to six weeks and sites can not contact the family by telephone, a second invitation letter will be mailed to the household (exception: California CADDRE site). At the Maryland CADDRE site, if no response card is received after the second set of mailings, a third invitation packet will be mailed at a later date (e.g., 6 months). This additional mailing will be identical to the first invitation packet described above. If no response is received after the third mailing, no further contact will be attempted.
If the invitee returns the response card indicating interest, recruitment proceeds with the invitation telephone call. In addition, if a source allows study staff to contact the potential participants by telephone prior to receiving a positive response from the response card, sites proceed with the invitation phone call after the first introductory packet.
The invitation phone call includes verbal consent for the SCQ screen and the study, eligibility screen, and the SCQ autism screen (Appendix J). Scoring of the SCQ occurs during the call. If the SCQ score falls into the range indicating autism, or the child has a previous autism diagnosis, the participant is invited during this call to participate in the full study. Of participants whose SCQ scores do not indicate autism and they do not have a previous autism diagnosis, only a sample may be invited into the study. Depending on the sampling process, individuals who are selected may need to be re-contacted with a second phone call to be invited into the study. The script used during the telephone call is in Appendix N.
If a participating primary caregiver is not the biological mother, the caregiver is asked to provide the biological mother’s contact information. The biological mother is contacted by a similar Invitation Phone Call, omitting the eligibility and developmental screening. If she consents to participate, she is contacted for only the data collection items that the primary caregiver is unable to complete. In addition, the participant is asked if the biological father resides in the same household. If he does not reside in the household, the caregiver is also asked to provide the biological father’s contact information. If provided, the biological father is contacted by phone and invited to complete the paternal medical history form, a buccal swab kit, and to come in for a blood draw. The biological father is only considered essential to the study if he is the primary caregiver (participants without biological father data are considered as having a complete data set).
The Enrollment Packet (Appendix E) is mailed to participants who verbally agree to be in the study. This packet is includes a cover letter (Appendix E.1), study flow diagram (Appendix E.1), consent documents to review (Appendix E.3), prep guides for the remaining data collection components (Appendices E.4 and E.5), a checklist of questionnaires (E.23) HIPAA medical records release forms (Appendix E.18), and a cheek cell collection kit (Appendix E.19-E.21). Sites also include a picture story (Appendices E.22) in the enrollment packet. This explains what the child will do during the clinic visit and makes them feel more comfortable with the process.
The general flow of the data collection begins with the enrollment packet. Please refer to Appendix D for a study flow diagram and a data collection instruments summary table. Ideally, the Caregiver Interview (Appendix B) occurs prior to the clinic visit. The medical record abstraction portion (Appendix S) of the study is completed by study staff independently of other study components.
Each check swab kit is assembled based on the availability of targeted participants in the household (e.g., a household including the biologic mother and child, but not the biologic father, would receive a kit including only consent forms and brushes for mother and child). Buccal cell sampling kits are mailed to multiple households, as needed, e.g. if
biological parents live in separate households from the index child. Kits include an instruction sheet (Appendix E.19) and a consent form (Appendix E.20).
Participants are asked to return the kit with a labeled Federal Express mailer provided by staff. Buccal cell samples are sent directly by the participant to the study Central Lab where they are stored at -80ºC until DNA is extracted. Participants whose samples are not received by the time of the clinic visit are given an opportunity to complete the sample at the time of the clinic visit.
Study staff call the participant one week after the enrollment packet has been mailed to answer questions about the packet, explain and schedule the caregiver interview, and to schedule the next data collection step – the questionnaire packets.
Participants complete questionnaire packets with assistance from study staff at a clinic or home visit or over the telephone. We allow participants to choose the option that works best for them. In addition, we allow participants to complete the packets as self-administered questionnaires. Assisting participants with the completion of the packets ensures that the forms are completed fully, allows staff to answer questions, and builds rapport with participants.
The First Questionnaire Packet includes the Paternal and Maternal Medical Histories (E.13-14), the Autoimmune Disease Survey (E.9), the GI Questionnaire (E.12), and the Paternal Occupational Questionnaire (E.15). Case participants also complete the Services and Treatments Questionnaire (F.3) and the Early Development Questionnaire (F.2). The Second Questionnaire Packet includes the Child Behavior Checklist (E.11), the Carey Temperament Scales (E.10), the Child Sleep Habits Questionnaire (E.16), and the Social Responsiveness Scales (E.17).
Staff call the participants after the First and Second Questionnaire Packet are complete to answer any questions and to schedule the clinic visits. If the participant has opted to complete the first packet as self-administered questionnaires, the staff member checks on the participant’s progress and answer any questions.
All primary caregivers of case and comparison children are asked to complete a caregiver interview by telephone. If the primary caregiver is not the biological mother, they are only asked questions related to sociodemographic factors and the child’s postnatal medical and developmental history (Sections A, B, and H of the caregiver interview (Appendix C2). Sites use their own staff to conduct the CATI.
The clinic visit includes four main components that can be split into different clinic visits or combined for one longer visit. Ideally, subcohort and NIC members are scheduled for one clinic visit. The Case group often requires more than one face-to-face visit, as the data collection is lengthier for this group.
The clinic visit component consists of four elements: 1) Intake, 2) Child Clinical, 3) Parent Biosample, and 4) Parent Interview. During Intake, study staff answer questions related to the study, review the self-administered questionnaires and HIPAA release forms, and consent the participant. The child clinical visit includes three components: a developmental assessment battery (Appendix G), a physical examination of the child (Appendix P), and a child blood and hair sample. The developmental battery includes the Mullen Scales of Early Learning (Appendix G2) which all children complete, and the ADOS (Appendix G1), which only case children complete.
Biological mothers and fathers in all three study groups (Case, NIC, and the subcohort) are asked to provide venous blood samples. Blood tubes are retained at appropriate temperature and shipped via Federal Express to the Central Lab the same day they are drawn. Filter paper cards are shipped to the Central Lab within seven days of collection. Please see Appendix R.1 for a more detailed description of the handling, shipment, and storage of these blood samples.
All primary caregivers of case children are asked to complete an interview asking questions about their child’s development. This interview includes 2 separate components: the ADI-R and the Vineland. Please see Appendix F for the parent interview.
The packet called self-administered Packet II (Appendix H), includes the Diet and Stool Diaries, is provided to the participant during the clinical visit. During the clinical visit, study staff provide the caregiver with a brief explanation for how to complete these forms and provide each participant with a packet to be returned to the study.
Medical record abstraction includes the maternal prenatal and, labor and delivery records as well as the records from other medical providers (i.e., gynecologist, allergists, psychiatrists) the biological mother might have visited three years prior to the child’s date of birth; and the neonatal and pediatric records of the child including any specialty providers (i.e, developmental pediatrician, allergist, gastroenterologist, psychiatrist) the child might have visited since birth. After the HIPAA medical records release authorization forms (Appendix E.18) have been received, medical record abstractors will contact relevant sources (clinics, hospitals) in order to obtain access to the medical records of participants. The Medical Records Request Script and Fax letter are Appendix S.1. All data will be collected electronically using the CIS. The CIS screens will be based on hard copy forms developed by the Data Collection Instruments Working Group (hard copies can be found in Appendix S).
B.3.Methods to Maximize Response Rates and Deal with Non-response
We are achieving a response rate across data collection items among enrolled participants of 50%-95%, and are targeting a 70% rate across all instruments. We have several follow-up calls and face-to-face visits to build rapport with the participant and to keep abreast of the participant’s progress and needs. This strategy facilitates the participant’s completion of the data collection protocol, and it also enhances the quality of data obtained from each participant. In the first and all subsequent contacts with the participant, we emphasize that we will work with the participant to help them complete the data collection process – in essence, a tailored approach. If the proposed data collection plan needs adjustment to fit the individual participant’s needs, it is done.
As described previously, all letters of invitation include a description of the study and other information to facilitate the respondent’s understanding of the project. If the respondent has additional questions about the study they are encouraged to call the Principal Investigator. The respondent is asked to return a response card indicating whether or not they are interested in participating in the study.
If there is no response from the invitee within 2 weeks, follow-up phone calls may be made by SEED staff in order to contact the respondent and explain the purpose of the study, answer questions and encourage participation. At least 9 attempts at phone contact are made; preferably on different days and different times of the day. For the first call, if the caregiver is not there, we will not leave a message. If on or after the 2nd call, we have not been able to reach the caregiver, we will leave a generic message asking the caregiver to call back. We will leave up to three messages for the caregiver. If after 9 attempts, the potential participant is not able to be contacted, no further attempts to contact will be made.
Verification of the telephone number and address information is made using telephone and crisscross directories.
All families will be provided incentives for the effort it takes to complete the study and costs incurred by the participant. Please see Section 9 (Payments to Respondents) for a detailed description of the incentive schedule.
Families may refuse participation in one component of the study and still be considered a study participant. We hope to enhance overall participation by not insisting that the family consent to all components of the study; although all participants will be encouraged to complete the entire study protocol. Also, study staff are trained in approaches to alleviate concerns about participation in order to increase the likelihood of obtaining consent.
SEED also implemented extensive public relation campaigns in each of the study catchment areas. These campaigns consisted of media spots via television, radio, and magazines/newsletters; promoting local physician awareness and knowledge of SEED; speaking at health care, service provider, advocacy and support group meetings; establishing community advisory boards, and other specific community activities that are appropriate to the potential study population. Several of our CA SEED staff were highlighted in a local news report demonstrating some of the techniques utilized during the clinical assessments. Several press releases were also published on the SEED study that was distributed locally at some of the SEED sites.
In the CHARGE Study, recruitment rates for control families with other developmental disabilities were significantly lower than those for the case families and the general population control families. In response, CHARGE created a second recruitment brochure that was less autism-centric and emphasized the study as looking at child development. These changes increased recruitment rates among non-case families (Culp, Personal Communication, April 6, 2007). SEED has used the CHARGE experience to develop a recruitment brochure that will be attractive to non-case families.
As described above, our population-based approach to subject ascertainment includes multiple clinical and education sources in each study area to identify potential CASE and NIC participants and birth certificates to identify potential subcohort participants. Some sites have access to demographic or address information of individuals in the pool of potential participants (e.g., from clinic or school lists or birth certificate information) prior to sending the invitation letter. Thus, these sites are able to assess differences in respondents and non-respondents to the invitation letter. In most cases, however, the sites do not have access to this information since they are not permitted access to personal information prior to positive response. In such instances, the sites are negotiating with the sources (clinic, school, or vital records office) to provide aggregate data on the pool of potential participants from that source so that the site can assess possible differences between the positive responses and the total pool of potential participants. The sites' ability to get these aggregate data, however, varies from source to source. Consequently, we do not expect that we will be able to systematically adjust for non-response to the invitation letter.
B.4. Tests of Procedures or Methods to be Undertaken
The maternal primary caregiver interview (Appendix C) was adapted from a questionnaire used for the National Birth Defects Prevention Study (OMB # 0920-0010, expiration 5/31/2009). In the process of modifying the questionnaire for SEED, some individual questions were adopted without any changes, but all sections were modified in some way to make the questionnaire more appropriate for this particular protocol. Additionally, investigators pilot tested the study instruments and materials, including recruitment methodology and materials, medical record ascertainment and abstraction, laboratory methods, self-administered questionnaires, and the caregiver interview.
B.5. Individuals Consulted on Statistical Aspects and Individuals Collection and/or Analyzing Data
The following individuals act as statistical consultants on the project.
Principal investigators at each study site:
Diana Schendel, Ph.D./Centers for Disease Control and Prevention, Atlanta, GA 404.498.3845, dcs6@cdc.gov
Lisa Miller, M.D./Colorado Department of Public Health and Environment 303.692.2663, lisa.miller@state.co.us
Lisa Croen, Ph.D., Kaiser Permanente Division of Research, 510.891.3463, lisa.a.croen@kp.org
Dani Fallin, Ph.D., Johns Hopkins University, 410-955-3463, dfallin@jhsph.eduJennifer Martin, Ph.D./University of Pennsylvania 215.898.4726, pinto@nursing.upenn.edu
Julie Daniels, Ph.D./University of North Carolina at Chapel Hill 919.966.7096, Julie_daniels@unc.edu
Co-investigators at each study site:
Gayle Windham, Ph.D., California Department of Health Services, Division of Environmental and Occupational Disease Control 510.620.3638, Gayle.Windham@cdph.ca.gov
Craig Newshaffer, Ph.D., Drexel University 215.762.7152, cnewscha@drexel.edu
Rebecca Landa, Ph.D., Kennedy Krieger Institute, 443.552.1000, landa@kennedykrieger.org
Susan Levy, M.D., Children’s Hospital of Philadelphia, 215.590.7528, levys@email.chop.edu
Cordelia Robinson, Ph.D., JFK Partners/UCHSC, 303.864.5261, robinson.cordelia@tchden.org
Laura Schieve, Ph.D./Centers for Disease Control and Prevention, Atlanta, GA 404-498-3888, ljs9@cdc.gov
Other CDC personnel that act as consultants for the GA SEED include:
Marshalyn Yeargin-Allsopp, M.D./Centers for Disease Control and Prevention, Atlanta, GA 404-498-3842, mxy1@cdc.gov
Catherine Rice, Ph.D./Centers for Disease Control and Prevention, Atlanta, GA 404-498-3847, cqr8@cdc.gov
Lisa Wiggins, PhD/ Centers for Disease Control and Prevention, Atlanta, GA 404-498-3875, LWiggins@cdc.gov
Each of the principal investigators and co-investigators helped design the overall study and data collection plan. These investigators have extensive experience in epidemiologic research concerning adverse reproductive and developmental outcomes, especially developmental disabilities, and have authored or co-authored numerous peer-reviewed publications in the epidemiology of developmental disabilities.
DCC personnel also act as statistical consultants. It is expected that these personnel will provide the necessary expertise in order to accomplish the DCC goals outlined in Section 3 (Use of Improved Information Technology).
References
Achenbach TM, Ruffle TM (2000). The Child Behavior Checklist and related forms for assessing behavioral/emotional problems and competencies. Pediatric Review Aug;21(8):265-71.
Achenbach, T. M., & Rescorla, L. A. (2000). Manual for ASEBA Preschool Forms & Profiles. Burlington, VT: University of Vermont, Research Center for Children, Youth, & Families.
Ackerman, T. F. (1989). An ethical framework for the practice of paying research subjects. IRB, 11(4), 1-4.
Bacchelli E, Maestrini E. 2006. Autism spectrum disorders: Molecular genetic advances. Am. J. Med. Genet. C. Semin. Med. Genet. 142 : 13-23
Barlow, W. E., Ichikawa, L., Rosner, D., & Izumi, S. (1999). Analysis of case-cohort designs. Journal of Clinical Epidemiology, 52(12), 1165-1172.
Burd, L, Severud, R, Kerbeshian, J, Klug, MG. Prenatal and perinatal risk factors for autism. Journal of Perinatal Medicine 1999; 27: 441-450.
Croen, L., Grether, J., Hoogstrate, J., Selvin, S. (2002). The changing prevalence of autism in California. Journal of Autism and Developmental Disorders, 32 (3), 207-215.
Culp, L. & Croen, L. (Personal Communication, November 27, 2006).
Culp, L. & Croen, L. (Personal Communication, April 6, 2007).
Dickert, N. & Grady, C. (1999). What’s the price of a research subject? Approaches to payment for research participation. New England Journal of Medicine, 341(3), 198-203.
Dunn, LB., Gordon, NE. (2005). Improving Informed Consent and Enhancing Recruitment for Research by Understanding Economic Behavior. JAMA, 293: 609-612.
Garcia-Closas, M., Egan, K. M., Abruzzo, J., Newcomb, P. A., Titus-Ernstoff, L., Franklin, T., Bender, P. K., Beck, J. C., Le Marchand, L., Lum, A., Alavanja, M., Hayes, R. B., Rutter, J., Buetow, K., Brinton, L. A., & Rothman, N. (2001). Collection of genomic DNA from adults in epidemiological studies by buccal cytobrush and mouthwash. Cancer Epidemiology, Biomarkers and Prevention, 10(6), 687-696.
Glasson EJ, Bower C, Petterson B, de Klerk N, Chaney G, Hallmayer JF. Perinatal factors and the development of autism. Arch Gen Psychiatry 2004; 61:618-627.
Groves, RM. (2006). Nonresponse rates and nonresponse bias in household surveys. Public Opinion Quarterly, 70: 646-675.
Hansen, E., Sullivan, N, Thurm, A. Ware, J., and Lord C. (2002) Social Communication Questionnaire (SCQ). International Meeting for Autism Research. Orlando, FL.
Harlow, S. & Linet, M. (1989) Agreement between questionnaire data and medical records: The evidence for accuracy of recall. American Journal of Epidemiology, 129(2): 233-248.
Hultman CM, Sparen P, Cnattingius S. Perinatal risk factors for infantile autism. Epidemiology 2002; 13: 417-422.
Juul-Dam N, Townsend J, Courchesne E. Prenatal, perinatal, and neonatal factors in
autism, pervasive developmental disorder-not otherwise specified, and the general population. Pediatrics 2001; 107.
Kaaks, R., van der Tweel, I., van Noord, P. A., & Riboli, E. (1994). Efficient use of biological banks for biochemical epidemiology: Exploratory hypothesis testing by means of a sequential t-test. Epidemiology, 5(4), 429-438
Klauck SM. 2006. Genetics of autism spectrum disorder. Eur. J. Hum. Genet. 14 : 714-20
Larsson HJ, Eaton WW, Madsen KM, Vestergaard M, Olesen AV, et al. 2005. Risk factors for autism: Perinatal factors, parental psychiatric history, and socioeconomic status. Am. J. Epidemiol. 161 : 916-25
Lord C.,& Risi S. (unpublished). Unpublished draft of the Diagnostic Criteria for CPEA.
Lord,
C., Risi, S., Lambrecht, L., Cook, E., Leventhal, B., DiLavore, P.,
Pickles, A., & Rutter, M. (2000). The Autism Diagnostic
Observation Schedule-Generic: A standard measure of social and
communication deficits associated with the spectrum of autism.
Journal of Autism and Developmental Disorders, 30, 205-223.
Lord, C., Rutter, M., & Le Couteur, A. (1994). Autism Diagnostic Interview-Revised: A revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. Journal of Autism and Developmental Disorders, 24, 659-685.
Lord,
C., Rutter, M., Goode, S., Heemsbergen, J., Jordan, H., Mawhood, L.,
& Schopler, E. (1989). Autism Diagnostic Observation Schedule: A
standardized observation of communicative and social behavior.
Journal of Autism and Developmental Disorders, 19, 185.
Macklin, R. (1981). “Due” and “undue” inducements: On paying money to research subjects. IRB, 3, 1-6.
McGee, G. (1997). Subject to payment? Journal of the American Medical Association, 278, 199-200.
Newschaffer, Craig & Lee, Nora. Personal Communication, December, 2003.
Newschaffer CJ, for SEED Investigators. The epidemiology of autism spectrum disorders. Annual Review of Public Health (in press)
Prentice, R. L. (1986). A case-cohort design for epidemiologic cohort studies and disease prevention trials. Biometrika, 73, 1-11.
Rice, C., Schendel, D., Cunniff, C., & Doernberg N. (2003). Public health monitoring of developmental disabilities with a focus on the autism spectrum disorders. American Journal of Medical Genetics (Sem in Med Gen 00:1-6).
Rockenbauer, M., Olsen, J., Czeizel, A.E., Pedersen, L., Sorensen, H.T., & EuroMAP Group. (2002) Recall bias in a case-control surveillance system on the use of medicine during pregnancy. Epidemiology,12: 461-466.
Rutter M. Aetiology of autism: findings and questions. Journal of Intellectual Disability Research 2005; 49:231-238.
Sato, T. (1992). Estimation of a common risk ratio in stratified case-cohort studies. Statistics in Medicine, 11, 1599-1605.
Singer, E., Bossarte, RM. (2006). Incentives for survey participation: are they coercive?. Am J Prev Med, 31: 411-418.
Singer, E., Van Hoewyk, J., Gebler, N., Raghunathan, T., McGonagle, K. (1999). The effect of incentives on response rates in interviewer-mediated surveys. Journal of Official Statistics, 15 (2): 217-230.
Tilley, B., Barnes, A., Bergstrahl, E., Labarthe, D., Noller, K., Colton, T., Adam, E. (1985) A comparison of pregnancy history recall and medical records: Implications for retrospective studies. American Journal of Epidemiology, 121 (2); 269-281
Tomeo, C., Rich-Edwards, J.W., Michels, K., Berkey, C., Hunter, D.J., Frazier, A., Willet, W., Buka, S. (1999) Reproducibility and validity of maternal recall of pregnancy-related events. Epidemiology, 10 (6): 774-777.
Wacholder, S., & Boivin, J. F. (1987). External comparisons with the case-cohort design. American Journal of Epidemiology, 126, 1198-1209.
Weinberg, C. R., Wilcox, A. J., Lie, R. T. (1998). A log-linear approach to case-parent triad data: Assessing effects of disease genes that act either directly or through maternal effects and that may be subject to parental imprinting. American Journal of Human Genetics, 62, 969-978.
Yu J, et al. (1983). A quantitative review of research design effects on response rates to questionnaires. Journal of Marketing Research, 20:36-44.
-
| File Type | application/msword |
| Author | sic3 |
| Last Modified By | Thelma Elaine Sims |
| File Modified | 2010-04-07 |
| File Created | 2010-04-07 |
© 2026 OMB.report | Privacy Policy